
High-Fat Diet Impairs Muscle Function and Increases the Risk of Environmental Heatstroke in Mice

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
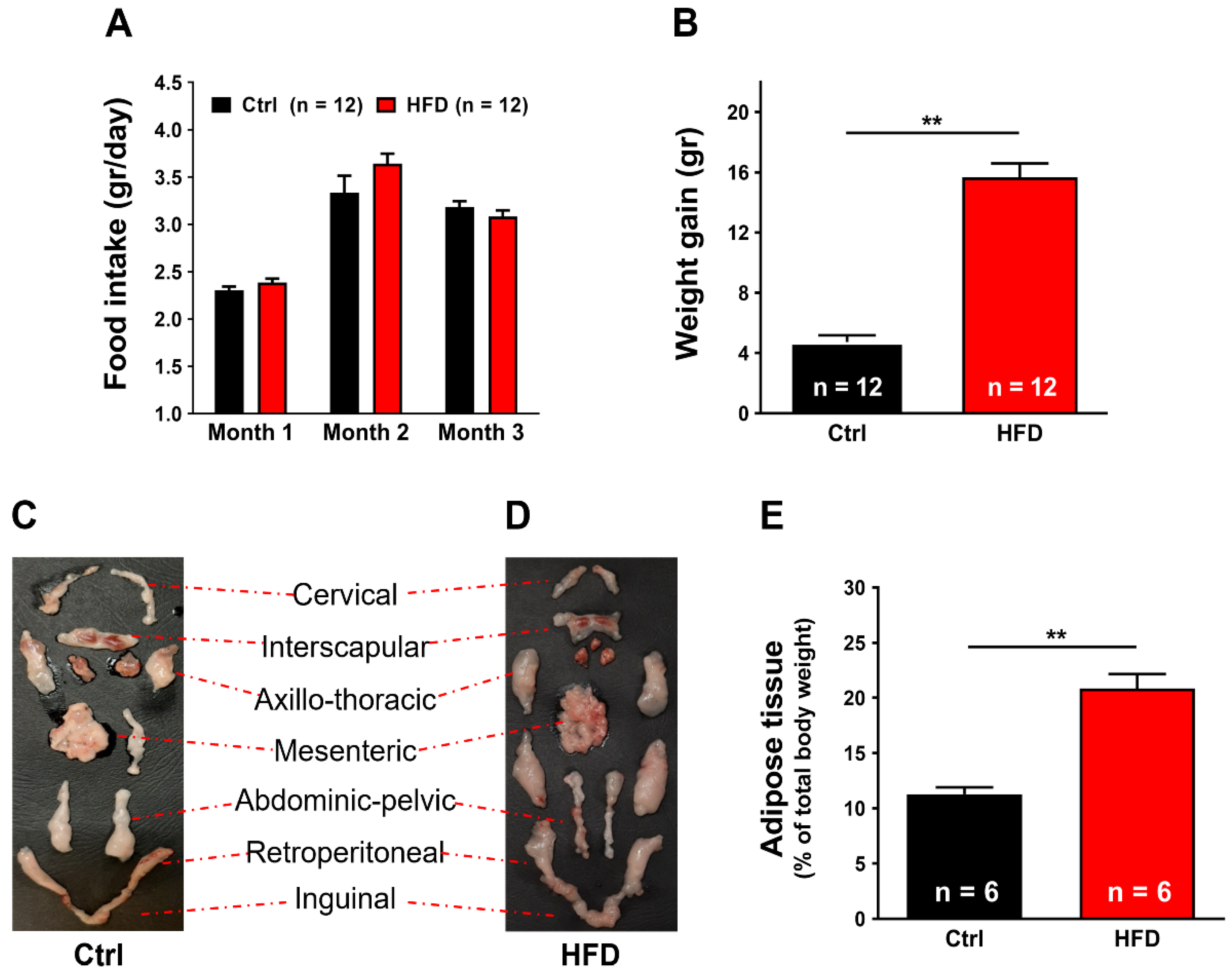
2.1. High-Fat Diet (HFD) Induced Metabolic Modifications in Adult WT Mice
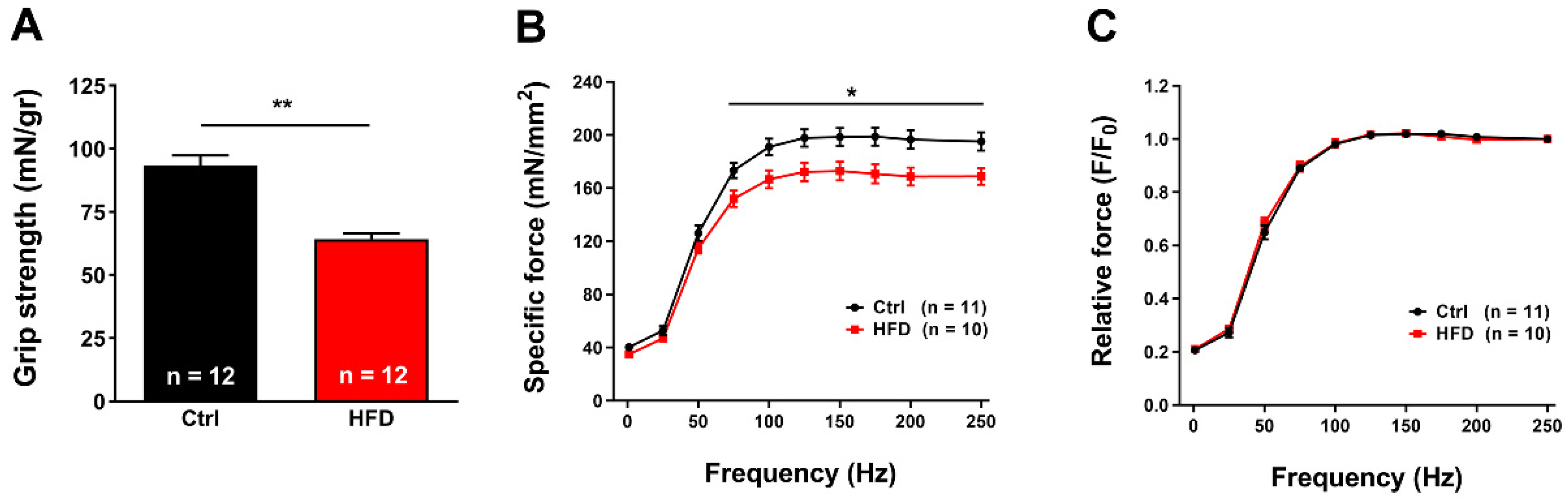
2.2. HFD Caused Muscle Dysfunction
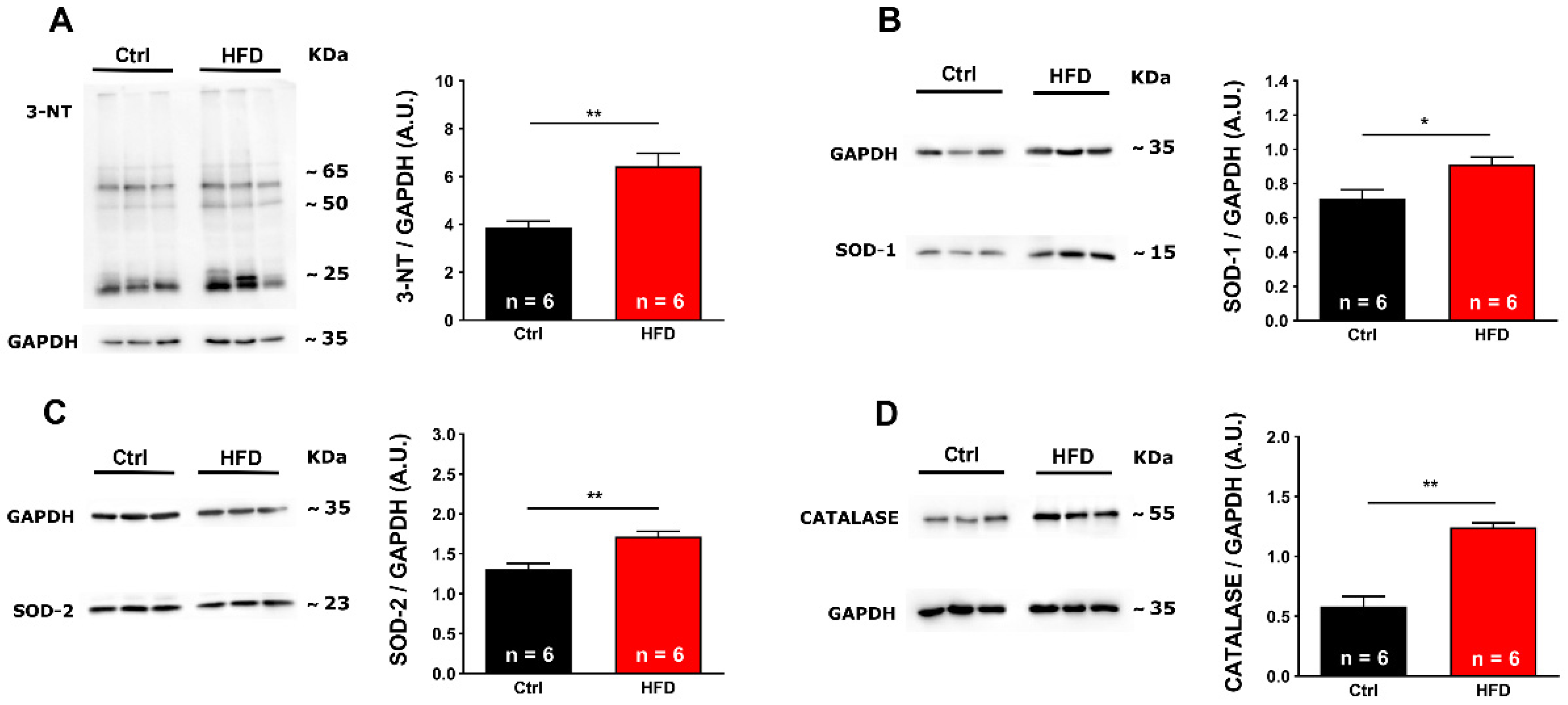
2.3. Oxidative Stress Was Elevated in Muscles from HFD Mice
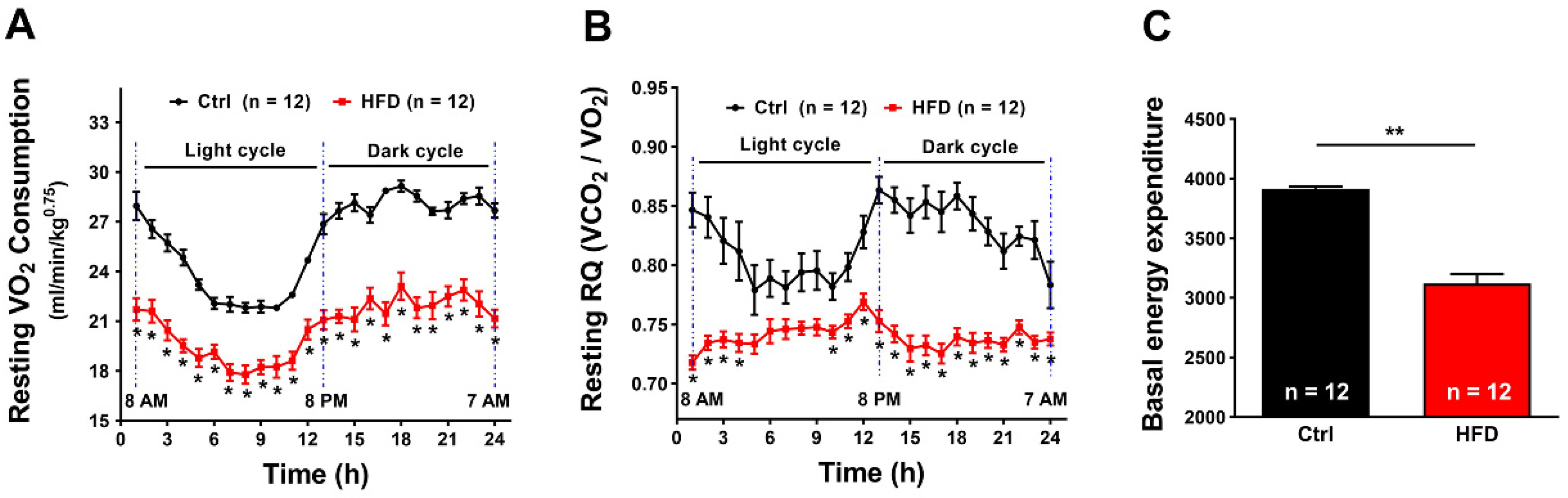
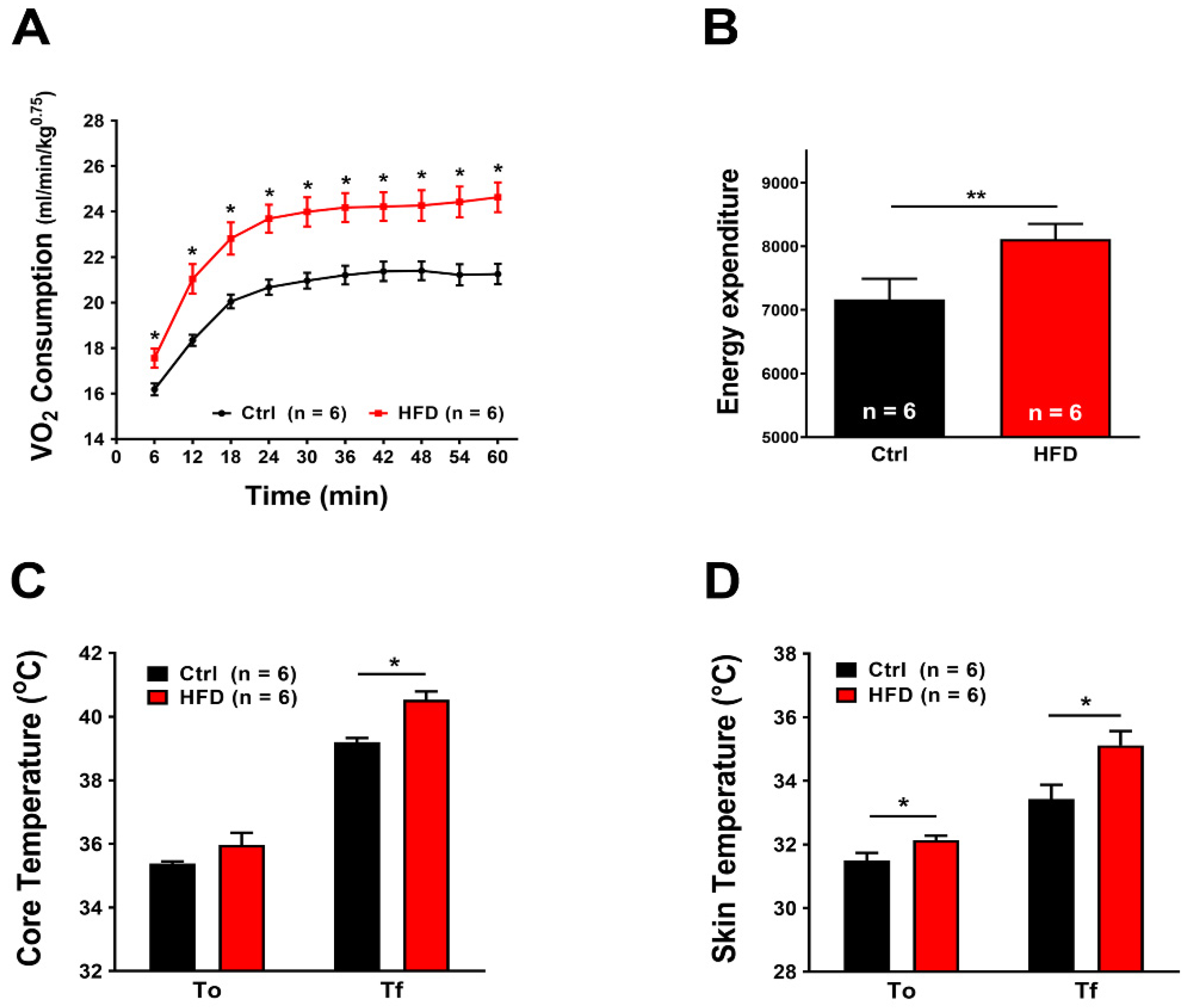
2.4. HFD Induced Increased Metabolic Rate during Heat Stress
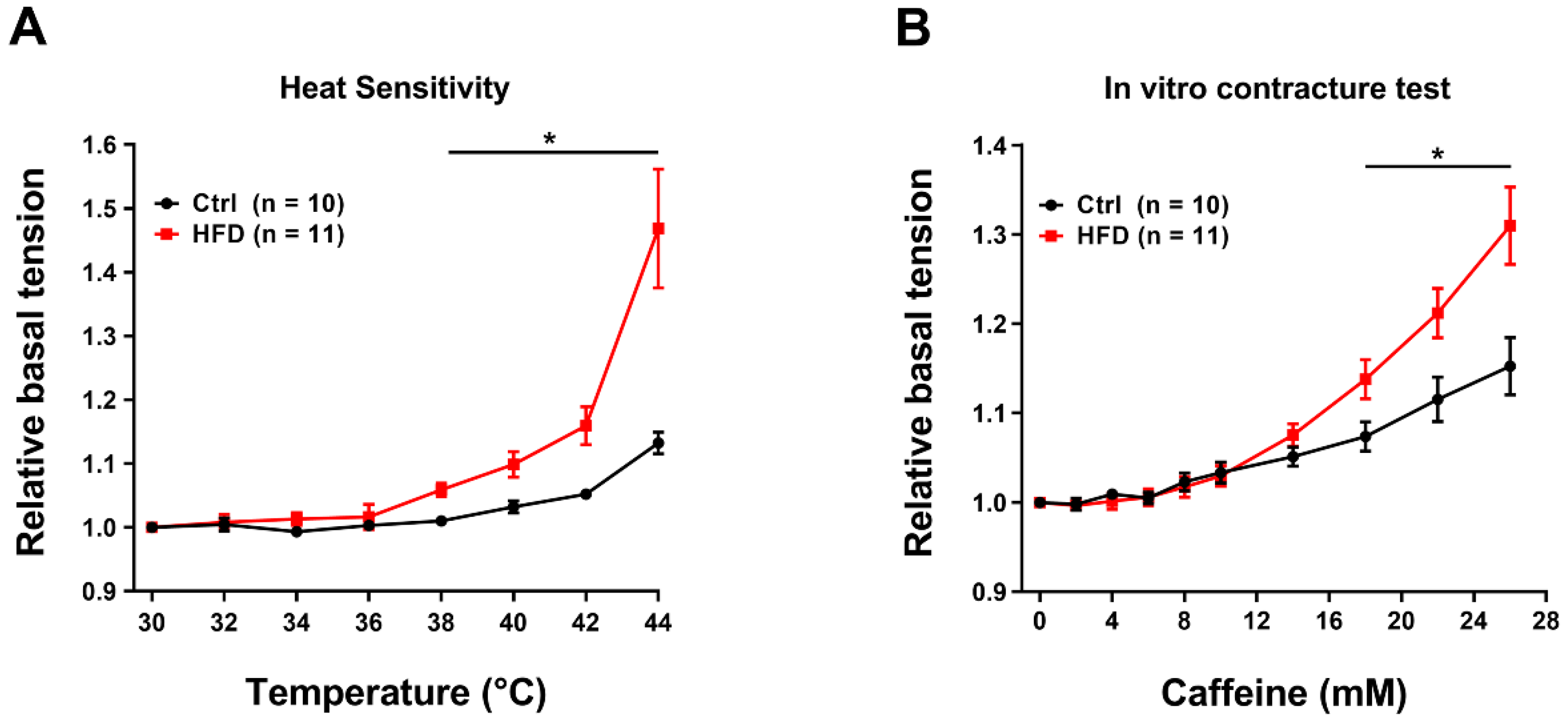
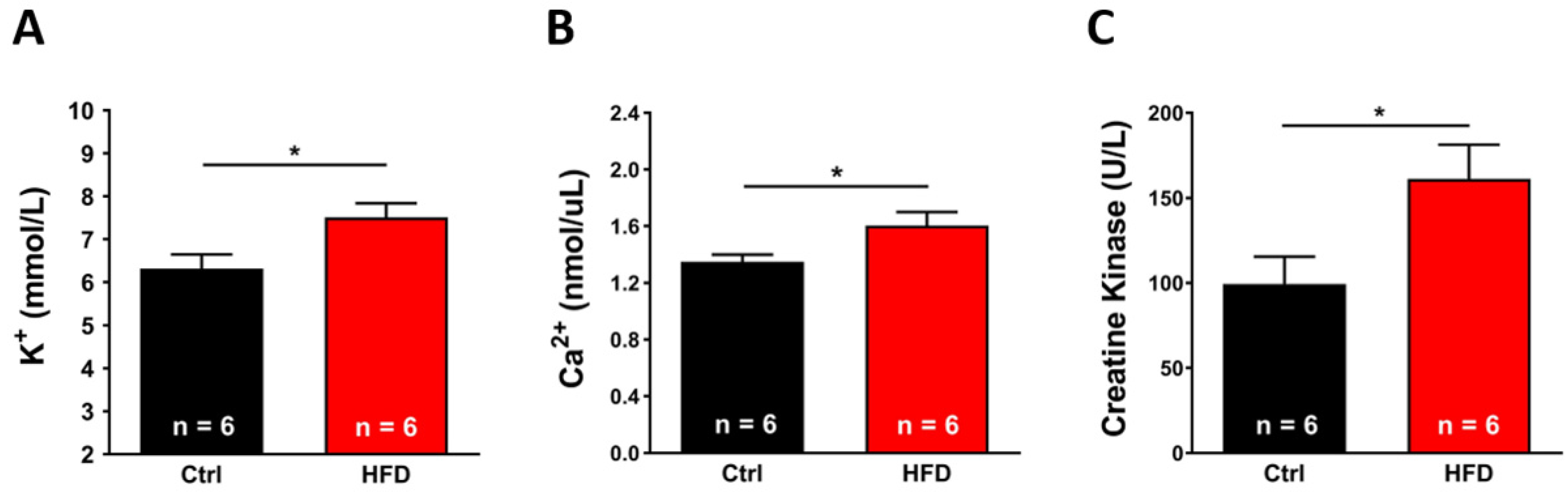
2.5. HFD Induced Hypersensitivity of Muscles to IVCT and Increased Levels of Markers of Muscle Damage
3. Discussion
3.1. State of the Art
3.2. Main Findings
3.3. Changes in Basal Metabolism, Oxidative Stress, and Muscle Dysfunction Caused by HFD
3.4. Increased Susceptibility to Heat Stress in HFD Mice
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. In Vivo Experiments
4.2.1. Grip Strength Test
4.2.2. Indirect Calorimetry
4.2.3. Heat Stress Protocol and Core Temperature Recording
4.3. Experiments after Animals Euthanasia
4.3.1. In Vitro Contracture Test (IVCT)
4.3.2. Blood Analysis
4.3.3. Assessment of Rhabdomyolysis
4.3.4. Measurements of Oxidative Stress
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gaffen, D.J.; Ross, R.J. Increased Summertime Heat Stress in the US. Nature 1998, 396, 529–530. [Google Scholar] [CrossRef]
- Basu, R.; Samet, J.M. Relation between Elevated Ambient Temperature and Mortality: A Review of the Epidemiologic Evidence. Epidemiol. Rev. 2002, 24, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Rossati, A. Global Warming and Its Health Impact. Int. J. Occup. Environ. Med. 2017, 8, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, C. Heat Wave. Weatherwise 1980, 33, 112–116. [Google Scholar] [CrossRef]
- Poumadère, M.; Mays, C.; le Mer, S.; Blong, R. The 2003 Heat Wave in France: Dangerous Climate Change Here and Now. Risk Anal. 2005, 25, 1483–1494. [Google Scholar] [CrossRef]
- Lim, C.L. Fundamental Concepts of Human Thermoregulation and Adaptation to Heat: A Review in the Context of Global Warming. Int. J. Environ. Res. Public Health 2020, 17, 7795. [Google Scholar] [CrossRef]
- Matthews, T.K.R.; Wilby, R.L.; Murphy, C. Communicating the Deadly Consequences of Global Warming for Human Heat Stress. Proc. Natl. Acad. Sci. USA 2017, 114, 3861–3866. [Google Scholar] [CrossRef] [Green Version]
- Yeo, T.P. Heat Stroke: A Comprehensive Review. AACN Clin. Issues 2004, 15, 280–293. [Google Scholar] [CrossRef]
- Bouchama, A.; Knochel, J.P. Heat Stroke. N. Engl. J. Med. 2002, 346, 1978–1988. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; McCarthy, T.; Lehmann-Horn, F. Genetics and Pathogenesis of Malignant Hyperthermia. Muscle Nerve 2000, 23, 4–17. [Google Scholar] [CrossRef]
- Lichtman, A.D.; Oribabor, C. Malignant Hyperthermia Following Systemic Rewarming after Hypothermic Cardiopulmonary Bypass. Anesth. Analg. 2006, 102, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Treves, S.; Anderson, A.A.; Ducreux, S.; Divet, A.; Bleunven, C.; Grasso, C.; Paesante, S.; Zorzato, F. Ryanodine Receptor 1 Mutations, Dysregulation of Calcium Homeostasis and Neuromuscular Disorders. Neuromuscul. Dis. 2005, 15, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, S.; Zorzato, F.; Müller, C.; Sewry, C.; Muntoni, F.; Quinlivan, R.; Restagno, G.; Girard, T.; Treves, S. Effect of Ryanodine Receptor Mutations on Interleukin-6 Release and Intracellular Calcium Homeostasis in Human Myotubes from Malignant Hyperthermia-Susceptible Individuals and Patients Affected by Central Core Disease. J. Biol. Chem. 2004, 279, 43838–43846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.M. Malignant Hyperthermia: Advances in Clinical Management and Diagnosis. Br. J. Anaesth. 2000, 85, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Hackl, W.; Winkler, M.; Mauritz, W.; Sporn, P.; Steinbereithner, K. Muscle Biopsy for Diagnosis of Malignant Hyperthermia Susceptibility in Two Patients with Severe Exercise-Induced Myolysis. Br. J. Anaesth. 1991, 66, 138–140. [Google Scholar] [CrossRef]
- Bendahan, D.; Kozak-Ribbens, G.; Confort-Gouny, S.; Ghattas, B.; Figarella-Branger, D.; Aubert, M.; Cozzone, P.J. A Noninvasive Investigation of Muscle Energetics Supports Similarities between Exertional Heat Stroke and Malignant Hyperthermia. Anesth. Analg. 2001, 93, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.W.; Nelson, T.E.; Anderson, I.L.; Kerr, D.D.; Burnap, T.K. Malignant Hyperthermia of Swine. Anesthesiology 1972, 36, 42–51. [Google Scholar] [CrossRef]
- Nelson, T.E.; Jones, E.W.; Venable, J.H.; Kerr, D.D. Malignant Hyperthermia of Poland China Swine: Studies of a Myogenic Etiology. Anesthesiology 1972, 36, 52–56. [Google Scholar] [CrossRef]
- Chelu, M.G.; Goonasekera, S.A.; Durham, W.J.; Tang, W.; Lueck, J.D.; Riehl, J.; Pessah, I.N.; Zhang, P.; Bhattacharjee, M.B.; Dirksen, R.T.; et al. Heat- and Anesthesia-Induced Malignant Hyperthermia in an RyR1 Knock-in Mouse. FASEB J. 2006, 20, 329–330. [Google Scholar] [CrossRef]
- Yang, T.; Riehl, J.; Esteve, E.; Matthaei, K.I.; Goth, S.; Allen, P.D.; Pessah, I.N.; Lopez, J.R. Pharmacologic and Functional Characterization of Malignant Hyperthermia in the R163C RyR1 Knock-in Mouse. Anesthesiology 2006, 105, 1164–1175. [Google Scholar] [CrossRef]
- Dainese, M.; Quarta, M.; Lyfenko, A.D.; Paolini, C.; Canato, M.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Anesthetic- and Heat-Induced Sudden Death in Calsequestrin-1-Knockout Mice. FASEB J. 2009, 23, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnier, F.A.; Michelucci, A.; Serano, M.; Pietrangelo, L.; Pecorai, C.; Boncompagni, S.; Protasi, F. Aerobic Training Prevents Heatstrokes in Calsequestrin-1 Knockout Mice by Reducing Oxidative Stress. Oxid. Med. Cell Longev. 2018, 2018, 4652480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Paolini, C.; Canato, M.; Wei-Lapierre, L.; Pietrangelo, L.; de Marco, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Antioxidants Protect Calsequestrin-1 Knockout Mice from Halothane- and Heat-Induced Sudden Death. Anesthesiology 2015, 123, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protasi, F.; Paolini, C.; Dainese, M. Calsequestrin-1: A New Candidate Gene for Malignant Hyperthermia and Exertional/Environmental Heat Stroke. J. Physiol. 2009, 587, 3095–3100. [Google Scholar] [CrossRef] [PubMed]
- Durham, W.J.; Aracena-Parks, P.; Long, C.; Rossi, A.E.; Goonasekera, S.A.; Boncompagni, S.; Galvan, D.L.; Gilman, C.P.; Baker, M.R.; Shirokova, N.; et al. RyR1 S-Nitrosylation Underlies Environmental Heat Stroke and Sudden Death in Y522S RyR1 Knockin Mice. Cell 2008, 133, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Estrogens Protect Calsequestrin-1 Knockout Mice from Lethal Hyperthermic Episodes by Reducing Oxidative Stress in Muscle. Oxid. Med. Cell Longev. 2017, 2017, 6936897. [Google Scholar] [CrossRef] [Green Version]
- Milajerdi, A.; Saneei, P.; Larijani, B.; Esmaillzadeh, A. The Effect of Dietary Glycemic Index and Glycemic Load on Inflammatory Biomarkers: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Am. J. Clin. Nutr. 2018, 107, 593–606. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased Oxidative Stress Precedes the Onset of High-Fat Diet-Induced Insulin Resistance and Obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Paolini, C.; Quarta, M.; Wei-LaPierre, L.; Michelucci, A.; Nori, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Oxidative Stress, Mitochondrial Damage, and Cores in Muscle from Calsequestrin-1 Knockout Mice. Skelet. Muscle 2015, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Paolini, C.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Strenuous Exercise Triggers a Life-Threatening Response in Mice Susceptible to Malignant Hyperthermia. FASEB J. 2017, 31, 3649–3662. [Google Scholar] [CrossRef] [Green Version]
- Ogino, K.; Wang, D.H. Biomarkers of Oxidative/Nitrosative Stress: An Approach to Disease Prevention. Acta Med. Okayama 2007, 61, 181–189. [Google Scholar] [PubMed]
- McCord, J.M.; Fridovich, I. The Utility of Superoxide Dismutase in Studying Free Radical Reactions. I. Radicals Generated by the Interaction of Sulfite, Dimethyl Sulfoxide, and Oxygen. J. Biol. Chem. 1969, 244, 6056–6063. [Google Scholar] [CrossRef]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive Oxygen Species: Impact on Skeletal Muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar]
- EMH Group. A Protocol for the Investigation of Malignant Hyperthermia (MH) Susceptibility. Br. J. Anaesth. 1984, 56, 1267–1269. [Google Scholar] [CrossRef]
- Larach, M.G. Standardization of the Caffeine Halothane Muscle Contracture Test. North American Malignant Hyperthermia Group. Anesth. Analg. 1989, 69, 511–515. [Google Scholar] [CrossRef]
- Lopez, J.R.; Linares, N.; Pessah, I.N.; Allen, P.D. Enhanced Response to Caffeine and 4-Chloro-m-Cresol in Malignant Hyperthermia-Susceptible Muscle Is Related in Part to Chronically Elevated Resting [Ca2+]i. Am. J. Physiol. Cell Physiol. 2005, 288, C606–C612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegazzin, V.; Scutari, E.; Treves, S.; Zorzato, F. Chlorocresol, an Additive to Commercial Succinylcholine, Induces Contracture of Human Malignant Hyperthermia-Susceptible Muscles via Activation of the Ryanodine Receptor Ca2+ Channel. Anesthesiology 1996, 84, 1380–1385. [Google Scholar] [CrossRef]
- Allen, M.; Raper, S.; Mitchell, J. Climate Change. Uncertainty in the IPCC’s Third Assessment Report. Science 2001, 293, 430–433. [Google Scholar] [CrossRef]
- Basu, R. High Ambient Temperature and Mortality: A Review of Epidemiologic Studies from 2001 to 2008. Environ. Health 2009, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Conti, S.; Masocco, M.; Meli, P.; Minelli, G.; Palummeri, E.; Solimini, R.; Toccaceli, V.; Vichi, M. General and Specific Mortality among the Elderly during the 2003 Heat Wave in Genoa (Italy). Environ. Res. 2007, 103, 267–274. [Google Scholar] [CrossRef]
- Donaldson, G.C.; Keatinge, W.R.; Saunders, R.D. Cardiovascular Responses to Heat Stress and Their Adverse Consequences in Healthy and Vulnerable Human Populations. Int. J. Hyperth. 2003, 19, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ramon, M.; Zanobetti, A.; Cavanagh, D.P.; Schwartz, J. Extreme Temperatures and Mortality: Assessing Effect Modification by Personal Characteristics and Specific Cause of Death in a Multi-City Case-Only Analysis. Environ. Health Perspect. 2006, 114, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Bark, N. Deaths of Psychiatric Patients during Heat Waves. Psychiatr. Serv. 1998, 49, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Schifano, P.; Cappai, G.; de Sario, M.; Michelozzi, P.; Marino, C.; Bargagli, A.M.; Perucci, C.A. Susceptibility to Heat Wave-Related Mortality: A Follow-up Study of a Cohort of Elderly in Rome. Environ. Health 2009, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafoggia, M.; Forastiere, F.; Agostini, D.; Biggeri, A.; Bisanti, L.; Cadum, E.; Caranci, N.; de’ Donato, F.; de Lisio, S.; de Maria, M.; et al. Vulnerability to Heat-Related Mortality: A Multicity, Population-Based, Case-Crossover Analysis. Epidemiology 2006, 17, 315–323. [Google Scholar] [CrossRef]
- Schwartz, J. Who Is Sensitive to Extremes of Temperature? A Case-Only Analysis. Epidemiology 2005, 16, 67–72. [Google Scholar] [CrossRef]
- Even, P.C.; Nadkarni, N.A. Indirect Calorimetry in Laboratory Mice and Rats: Principles, Practical Considerations, Interpretation and Perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R459–R476. [Google Scholar] [CrossRef]
- Abbott, W.G.; Howard, B.V.; Ruotolo, G.; Ravussin, E. Energy Expenditure in Humans: Effects of Dietary Fat and Carbohydrate. Am. J. Physiol. 1990, 258, E347–E351. [Google Scholar] [CrossRef]
- Thomas, C.D.; Peters, J.C.; Reed, G.W.; Abumrad, N.N.; Sun, M.; Hill, J.O. Nutrient Balance and Energy Expenditure during Ad Libitum Feeding of High-Fat and High-Carbohydrate Diets in Humans. Am. J. Clin. Nutr. 1992, 55, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Tataranni, P.A.; Ravussin, E. Effect of Fat Intake on Energy Balance. Ann. N. Y. Acad. Sci. 1997, 819, 37–43. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Oxidation Enhances Myofibrillar Protein Degradation via Calpain and Caspase-3. Free Radic. Biol. Med. 2010, 49, 1152–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulle, S.; Protasi, F.; di Tano, G.; Pietrangelo, T.; Beltramin, A.; Boncompagni, S.; Vecchiet, L.; Fanò, G. The Contribution of Reactive Oxygen Species to Sarcopenia and Muscle Ageing. Exp. Gerontol. 2004, 39, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative Damage and Mitochondrial Decay in Aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roubenoff, R.; Hughes, V.A. Sarcopenia: Current Concepts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, M716–M724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, W.J. What Is Sarcopenia? J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 5–8. [Google Scholar] [CrossRef]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-Dependent Uncoupling of Mitochondria from Ca2(+) Release Units in Skeletal Muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef] [Green Version]
- Alberts, P.; Johansson, B.G.; McArthur, R.A. Characterization of Energy Expenditure in Rodents by Indirect Calorimetry. Curr. Protoc. Neurosci. 2006, 36, 9–23. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serano, M.; Paolini, C.; Michelucci, A.; Pietrangelo, L.; Guarnier, F.A.; Protasi, F. High-Fat Diet Impairs Muscle Function and Increases the Risk of Environmental Heatstroke in Mice. Int. J. Mol. Sci. 2022, 23, 5286. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23095286
Serano M, Paolini C, Michelucci A, Pietrangelo L, Guarnier FA, Protasi F. High-Fat Diet Impairs Muscle Function and Increases the Risk of Environmental Heatstroke in Mice. International Journal of Molecular Sciences. 2022; 23(9):5286. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23095286
Chicago/Turabian StyleSerano, Matteo, Cecilia Paolini, Antonio Michelucci, Laura Pietrangelo, Flavia A. Guarnier, and Feliciano Protasi. 2022. "High-Fat Diet Impairs Muscle Function and Increases the Risk of Environmental Heatstroke in Mice" International Journal of Molecular Sciences 23, no. 9: 5286. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23095286