
Sperm Energy Restriction and Recovery (SER) Alters Epigenetic Marks during the First Cell Cycle of Development in Mice


Abstract
:1. Introduction
2. Results
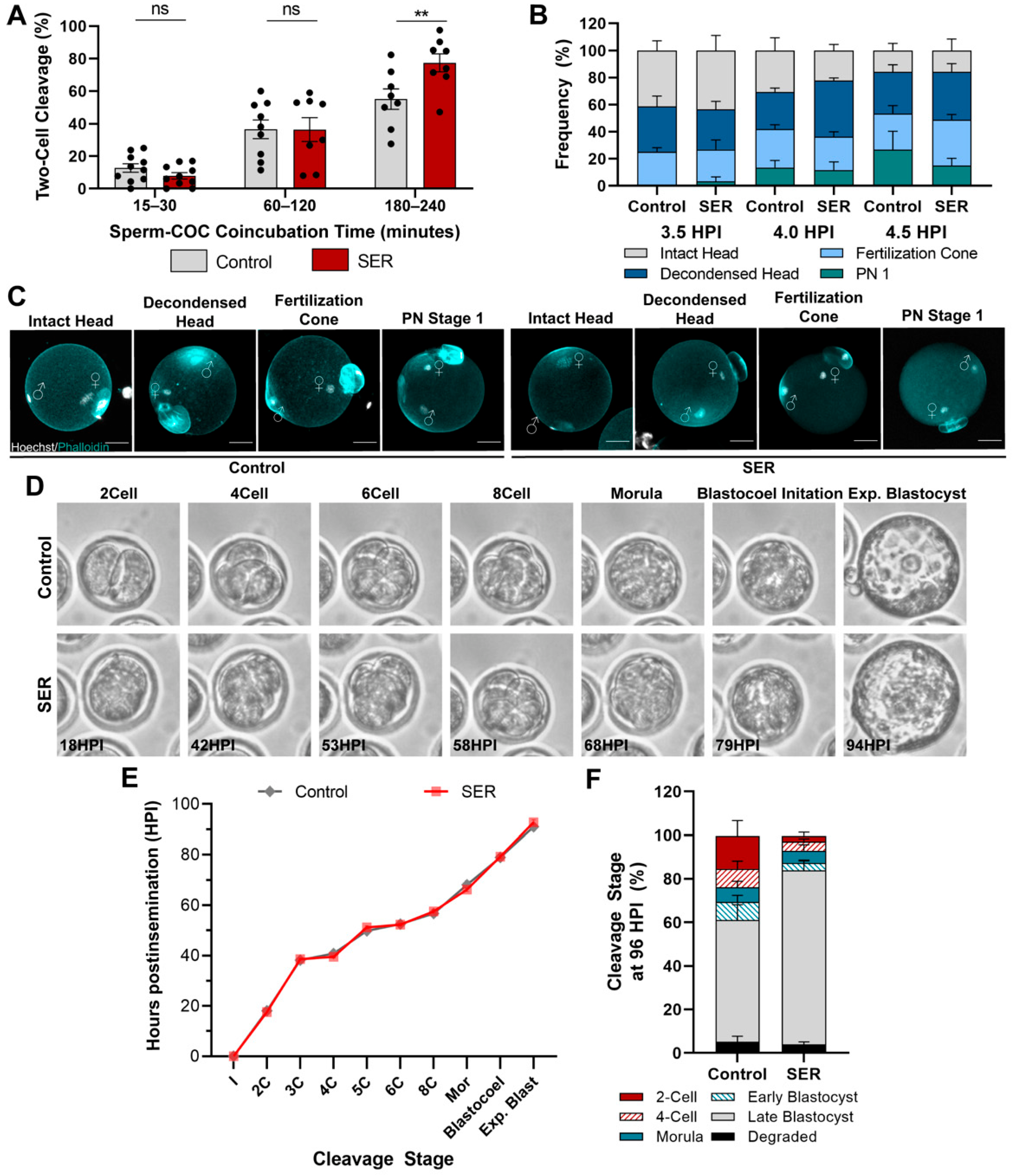
2.1. Timing of Fertilization and Early Embryo Cleavages Are Similar between SER- and Control-Generated Embryos
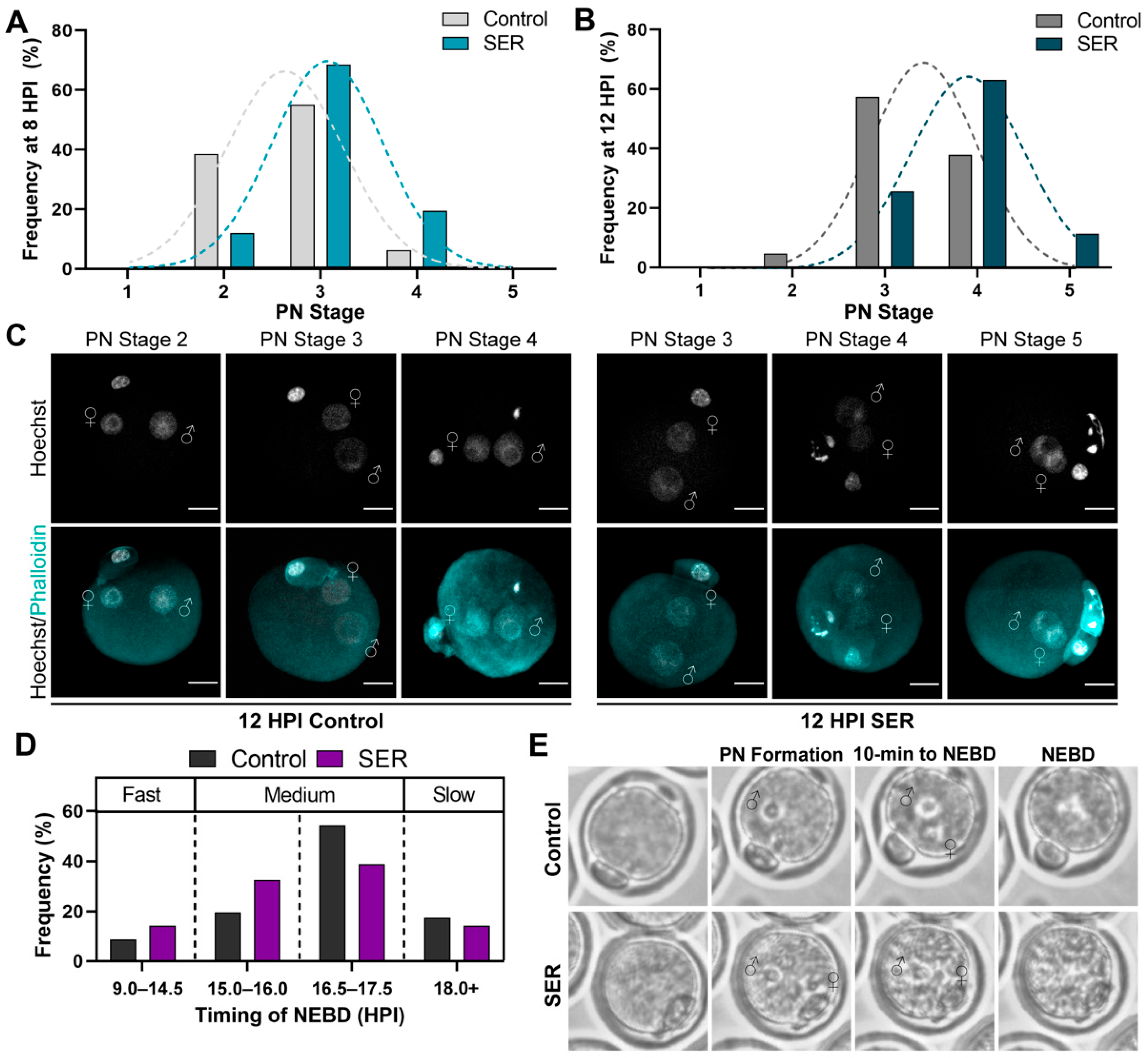
2.2. SER-Generated Embryos Are Advanced in Pronuclear Stage Progression
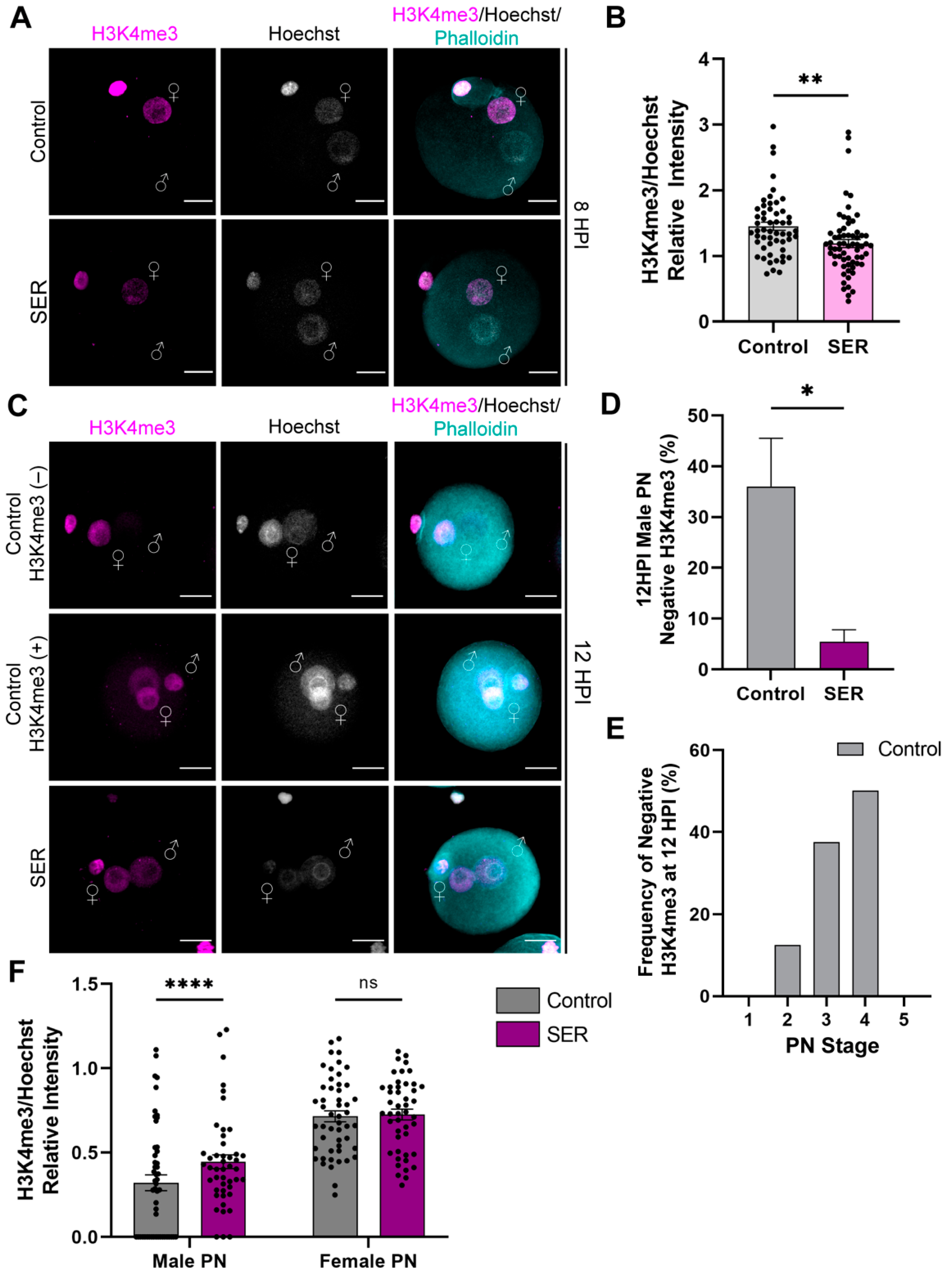
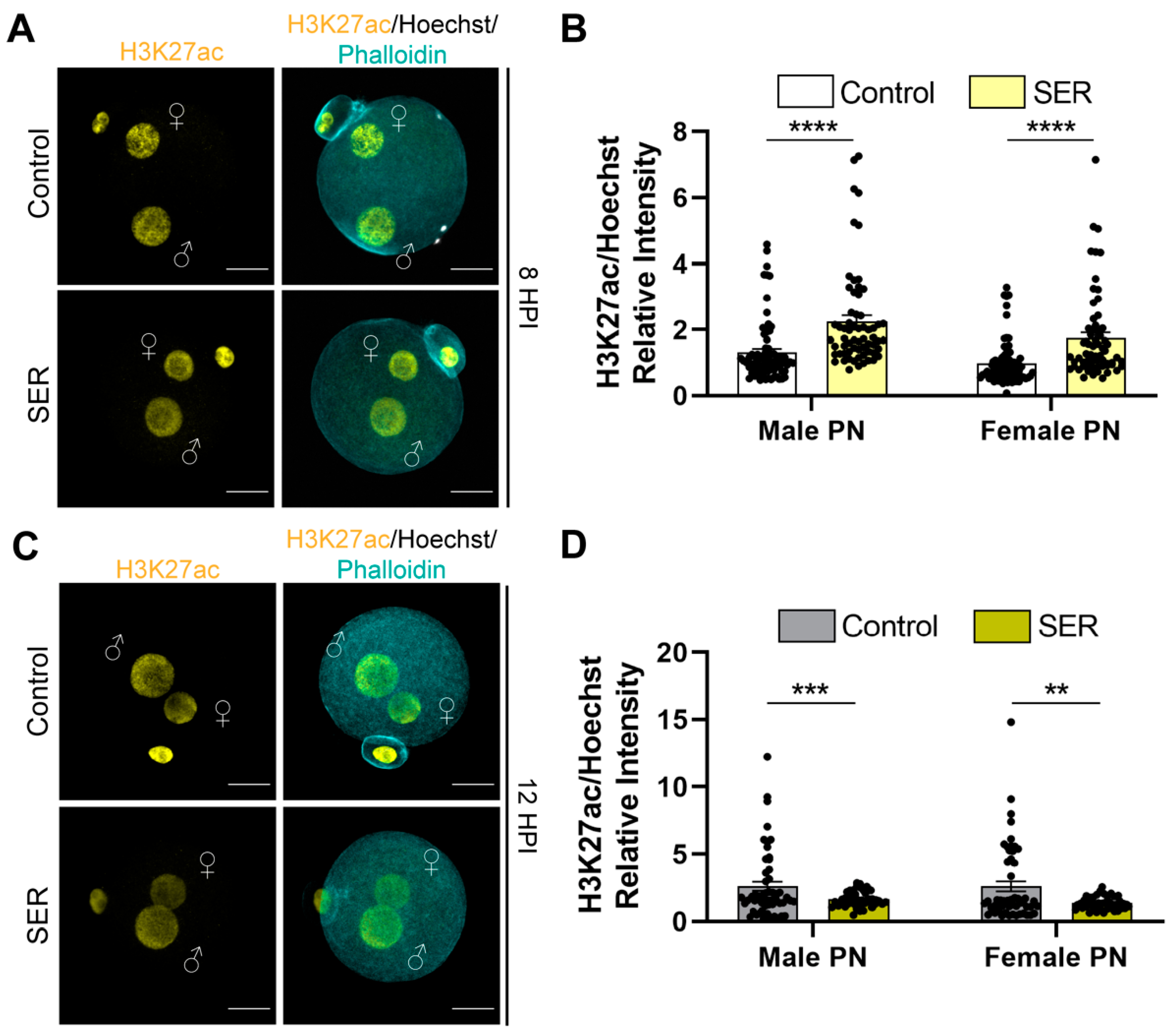
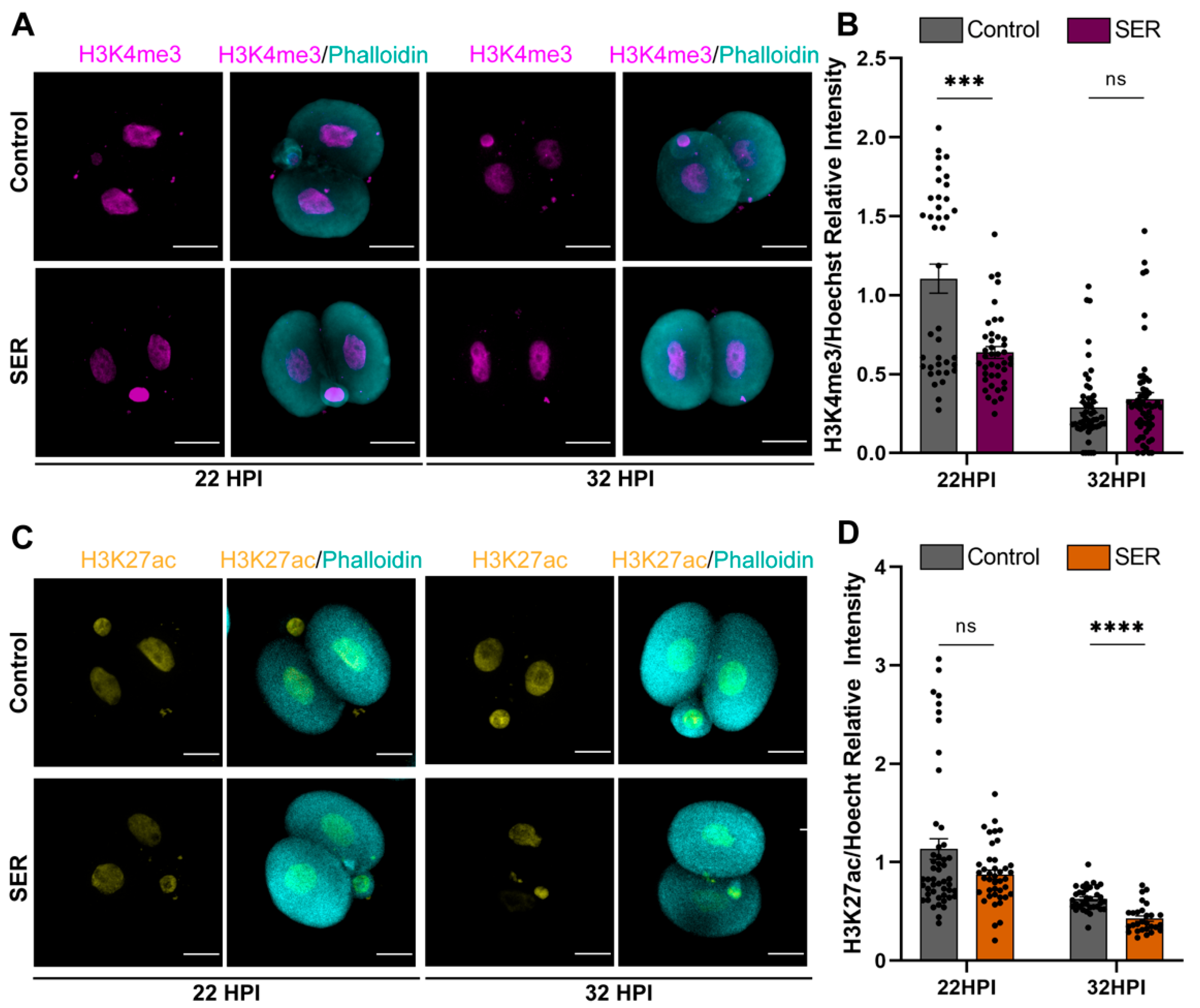
2.3. SER Results in Differences in Histone Modifications in the Male PN during the First Cell Cycle
2.4. Differences in Histone Modifications Continued to Be Observed after the First Cleavage
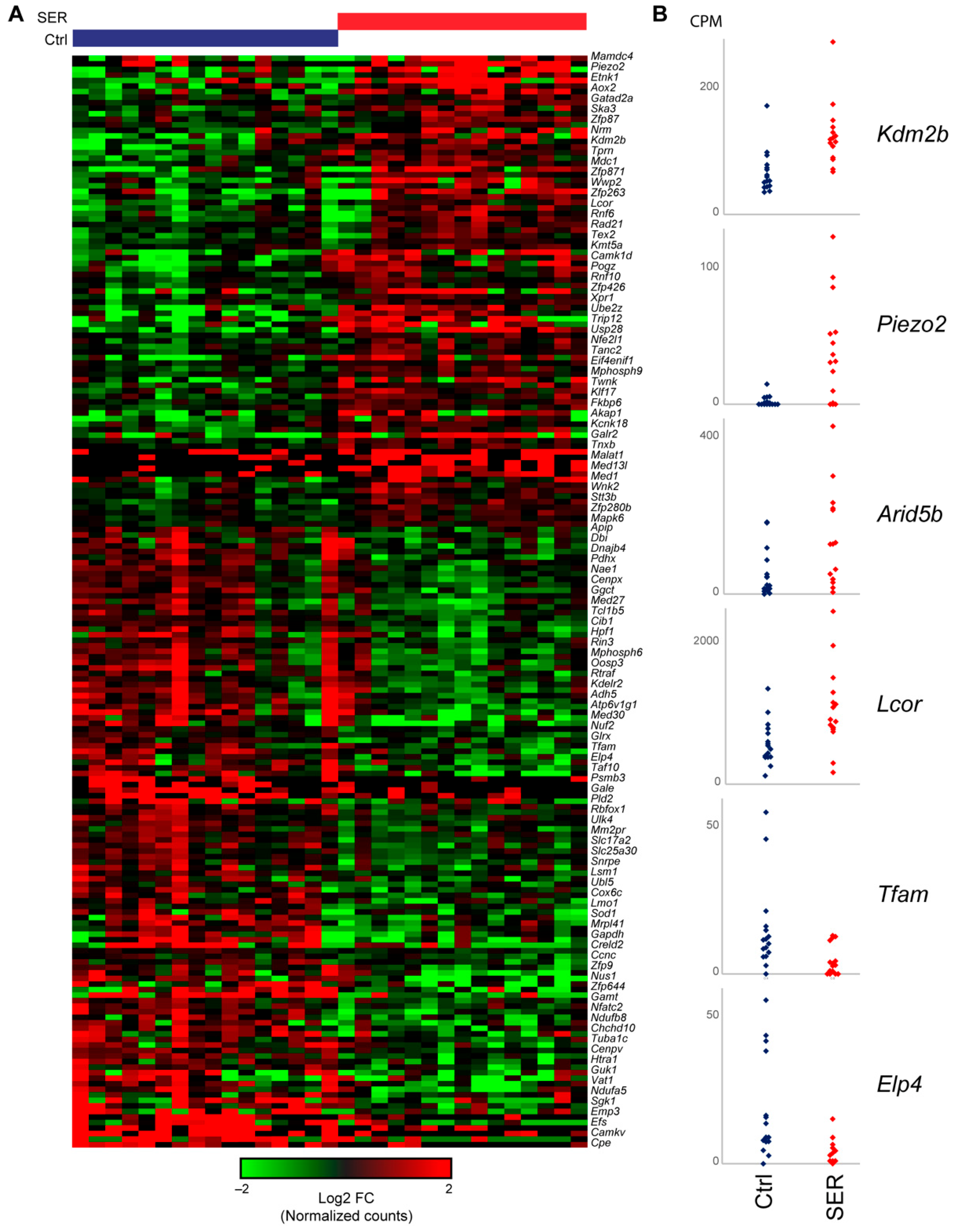
2.5. SER Treatment Results in Changes to Gene Expression in Late Two-Cell Embryos
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Media
4.3. SER Treatment
4.4. In Vitro Fertilization (IVF) and Embryo Culture
4.5. Sperm–COCs Coincubation Assay
4.6. Time-Lapse Imaging
4.7. Sperm Head Decondensation Assay
4.8. Immunofluorescence
4.9. Imaging Analysis
4.10. Single Embryo RNAseq
4.11. Analysis of Smart-seq3 Sequencing Data
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanders, J.R.; Jones, K.T. Regulation of the meiotic divisions of mammalian oocytes and eggs. Biochem. Soc. Trans. 2018, 46, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, J.G.; Kurokawa, M.; Fissore, R.A.; Schultz, R.M.; Williams, C.J. Transgenic RNA interference reveals role for mouse sperm phospholipase Czeta in triggering Ca2+ oscillations during fertilization. Biol. Reprod. 2005, 72, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, C.M.; Larman, M.G.; Parrington, J.; Cox, L.J.; Royse, J.; Blayney, L.M.; Swann, K.; Lai, F.A. PLC zeta: A sperm-specific trigger of Ca2+ oscillations in eggs and embryo development. Development 2002, 129, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Ducibella, T.; Fissore, R. The roles of Ca2+, downstream protein kinases, and oscillatory signaling in regulating fertilization and the activation of development. Dev. Biol. 2008, 315, 257–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutovsky, P.; Schatten, G. Paternal contributions to the mammalian zygote: Fertilization after sperm—Egg fusion. Int. Rev. Cytol. 2000, 195, 1–65. [Google Scholar]
- Scheffler, K.; Uraji, J.; Jentoft, I.; Cavazza, T.; Monnich, E.; Mogessie, B.; Schuh, M. Two mechanisms drive pronuclear migration in mouse zygotes. Nat. Commun. 2021, 12, 841. [Google Scholar] [CrossRef]
- Austin, C.R. Fertilization of mammalian eggs in vitro. Int. Rev. Cytol. 1961, 12, 337–359. [Google Scholar]
- Otsuki, J.; Iwasaki, T.; Tsuji, Y.; Katada, Y.; Sato, H.; Tsutsumi, Y.; Hatano, K.; Furuhashi, K.; Matsumoto, Y.; Kokeguchi, S.; et al. Potential of zygotes to produce live births can be identified by the size of the male and female pronuclei just before their membranes break down. Reprod. Med. Biol. 2017, 16, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Lu, X.; Dean, J. The maternal to zygotic transition in mammals. Mol. Aspects Med. 2013, 34, 919–938. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.I.; Funaya, S.; Tsukioka, D.; Kawamura, M.; Suzuki, Y.; Suzuki, M.G.; Schultz, R.M.; Aoki, F. Minor zygotic gene activation is essential for mouse preimplantation development. Proc. Natl. Acad. Sci. USA 2018, 115, E6780–E6788. [Google Scholar] [CrossRef] [Green Version]
- Aoki, F.; Worrad, D.M.; Schultz, R.M. Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo. Dev. Biol. 1997, 181, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asami, M.; Lam, B.Y.H.; Ma, M.K.; Rainbow, K.; Braun, S.; VerMilyea, M.D.; Yeo, G.S.H.; Perry, A.C.F. Human embryonic genome activation initiates at the one-cell stage. Cell Stem Cell 2021, 29, 209–216.e4. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.A.; Alda-Catalinas, C.; Reik, W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 2018, 19, 436–450. [Google Scholar] [CrossRef]
- Gamarra, N.; Narlikar, G.J. Collaboration through chromatin: Motors of transcription and chromatin structure. J. Mol. Biol. 2021, 433, 166876. [Google Scholar] [CrossRef] [PubMed]
- Marcho, C.; Cui, W.; Mager, J. Epigenetic dynamics during preimplantation development. Reproduction 2015, 150, R109-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Adenot, P.G.; Mercier, Y.; Renard, J.P.; Thompson, E.M. Differential H4 acetylation of paternal and maternal chromatin precedes DNA replication and differential transcriptional activity in pronuclei of 1-cell mouse embryos. Development 1997, 124, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.R.; Liu, Y.; Shang, M.B.; Yang, X.X.; Ding, B.; Gao, J.G.; Wang, R.; Li, W.Y. Differences in H3K4 trimethylation in in vivo and in vitro fertilization mouse preimplantation embryos. Genet. Mol. Res. 2012, 11, 1099–1108. [Google Scholar] [CrossRef]
- Comizzoli, P.; Wildt, D.E.; Pukazhenthi, B.S. In vitro development of domestic cat embryos following intra-cytoplasmic sperm injection with testicular spermatozoa. Theriogenology 2006, 66, 1659–1663. [Google Scholar] [CrossRef]
- Comizzoli, P.; Paulson, E.E.; McGinnis, L.K. The mutual benefits of research in wild animal species and human-assisted reproduction. J. Assist. Reprod. Genet. 2018, 35, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Patrizio, P.; Bianchi, V.; Lalioti, M.D.; Gerasimova, T.; Sakkas, D. High rate of biological loss in assisted reproduction: It is in the seed, not in the soil. Reprod. Biomed. Online 2007, 14, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Assisted Reproductive Technology Fertility Clinic and National Summary Report; US Department of Health and Human Services: Washington, DC, USA, 2021. [Google Scholar]
- European IVF Monitoring Consortium (EIM), for the European Society of Human Reproduction and Embryology (ESHRE). ART in Europe, 2018: Results generated from European registries by ESHRE. Hum. Reprod. Open 2022, 2022, hoac022. [Google Scholar]
- Navarrete, F.A.; Aguila, L.; Martin-Hidalgo, D.; Tourzani, D.A.; Luque, G.M.; Ardestani, G.; Garcia-Vazquez, F.A.; Levin, L.R.; Buck, J.; Darszon, A.; et al. Transient Sperm Starvation Improves the Outcome of Assisted Reproductive Technologies. Front. Cell Dev. Biol. 2019, 7, 262. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cardenas, C.; Romarowski, A.; Orta, G.; De la Vega-Beltran, J.L.; Martin-Hidalgo, D.; Hernandez-Cruz, A.; Visconti, P.E.; Darszon, A. Starvation induces an increase in intracellular calcium and potentiates the progesterone-induced mouse sperm acrosome reaction. FASEB J. 2021, 35, e21528. [Google Scholar] [CrossRef]
- Yang, F.; Gracia Gervasi, M.; Orta, G.; Tourzani, D.A.; De la Vega-Beltran, J.L.; Ruthel, G.; Darszon, A.; Visconti, P.E.; Wang, P.J. C2CD6 regulates targeting and organization of the CatSper calcium channel complex in sperm flagella. Development 2022, 149, dev199988. [Google Scholar] [CrossRef]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevicius, J.; Lukinavicius, G.; Elder, K.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860–2877.e22. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.A.; Jung, I.; Aanes, H.; Greggains, G.D.; Manaf, A.; Lerdrup, M.; Li, G.; Kuan, S.; Li, B.; Lee, A.Y.; et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 2016, 537, 548–552. [Google Scholar] [CrossRef]
- Castillo, J.; Jodar, M.; Oliva, R. The contribution of human sperm proteins to the development and epigenome of the preimplantation embryo. Hum. Reprod. Update 2018, 24, 535–555. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U. Paternal Contributions to Offspring Health: Role of Sperm Small RNAs in Intergenerational Transmission of Epigenetic Information. Front. Cell Dev. Biol. 2019, 7, 215. [Google Scholar] [CrossRef]
- Jung, Y.H.; Kremsky, I.; Gold, H.B.; Rowley, M.J.; Punyawai, K.; Buonanotte, A.; Lyu, X.; Bixler, B.J.; Chan, A.W.S.; Corces, V.G. Maintenance of CTCF- and Transcription Factor-Mediated Interactions from the Gametes to the Early Mouse Embryo. Mol. Cell 2019, 75, 154–171.e5. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, J.; Nijmeijer, B.; Hossain, M.J.; Eguren, M.; Schneider, I.; Politi, A.Z.; Roberti, M.J.; Hufnagel, L.; Hiiragi, T.; Ellenberg, J. Dual-spindle formation in zygotes keeps parental genomes apart in early mammalian embryos. Science 2018, 361, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matas, C.; Coy, P.; Romar, R.; Marco, M.; Gadea, J.; Ruiz, S. Effect of sperm preparation method on in vitro fertilization in pigs. Reproduction 2003, 125, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tesarik, J. Control of Maternal-to-Zygotic Transition in Human Embryos and Other Animal Species (Especially Mouse): Similarities and Differences. Int. J. Mol. Sci. 2022, 23, 8562. [Google Scholar] [CrossRef] [PubMed]
- McLay, D.W.; Clarke, H.J. Remodelling the paternal chromatin at fertilization in mammals. Reproduction 2003, 125, 625–633. [Google Scholar] [CrossRef]
- Abe, K.; Yamamoto, R.; Franke, V.; Cao, M.; Suzuki, Y.; Suzuki, M.G.; Vlahovicek, K.; Svoboda, P.; Schultz, R.M.; Aoki, F. The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3’ processing. EMBO J. 2015, 34, 1523–1537. [Google Scholar] [CrossRef] [Green Version]
- Wiekowski, M.; Miranda, M.; DePamphilis, M.L. Regulation of gene expression in preimplantation mouse embryos: Effects of the zygotic clock and the first mitosis on promoter and enhancer activities. Dev. Biol. 1991, 147, 403–414. [Google Scholar] [CrossRef]
- Majumder, S.; Miranda, M.; DePamphilis, M.L. Analysis of gene expression in mouse preimplantation embryos demonstrates that the primary role of enhancers is to relieve repression of promoters. EMBO J. 1993, 12, 1131–1140. [Google Scholar] [CrossRef]
- Funaya, S.; Aoki, F. Regulation of zygotic gene activation by chromatin structure and epigenetic factors. J. Reprod. Dev. 2017, 63, 359–363. [Google Scholar] [CrossRef] [Green Version]
- Santos, F.; Peters, A.H.; Otte, A.P.; Reik, W.; Dean, W. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 2005, 280, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepikhov, K.; Walter, J. Differential dynamics of histone H3 methylation at positions K4 and K9 in the mouse zygote. BMC Dev. Biol. 2004, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zheng, H.; Huang, B.; Li, W.; Xiang, Y.; Peng, X.; Ming, J.; Wu, X.; Zhang, Y.; Xu, Q.; et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 2016, 537, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Marinho, L.S.R.; Rissi, V.B.; Lindquist, A.G.; Seneda, M.M.; Bordignon, V. Acetylation and methylation profiles of H3K27 in porcine embryos cultured in vitro. Zygote 2017, 25, 575–582. [Google Scholar] [CrossRef]
- Kang, M.H.; You, S.Y.; Hong, K.; Kim, J.H. DMSO impairs the transcriptional program for maternal-to-embryonic transition by altering histone acetylation. Biomaterials 2020, 230, 119604. [Google Scholar] [CrossRef]
- Higuchi, C.; Shimizu, N.; Shin, S.W.; Morita, K.; Nagai, K.; Anzai, M.; Kato, H.; Mitani, T.; Yamagata, K.; Hosoi, Y.; et al. Ubiquitin-proteasome system modulates zygotic genome activation in early mouse embryos and influences full-term development. J. Reprod. Dev. 2018, 64, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, F.A.; Alvau, A.; Lee, H.C.; Levin, L.R.; Buck, J.; Leon, P.M.; Santi, C.M.; Krapf, D.; Mager, J.; Fissore, R.A.; et al. Transient exposure to calcium ionophore enables in vitro fertilization in sterile mouse models. Sci. Rep. 2016, 6, 33589. [Google Scholar] [CrossRef] [Green Version]
- Conine, C.C.; Sun, F.; Song, L.; Rivera-Perez, J.A.; Rando, O.J. Small RNAs Gained during Epididymal Transit of Sperm Are Essential for Embryonic Development in Mice. Dev. Cell 2018, 46, 470–480.e3. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F.; et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.H.; Sauria, M.E.G.; Lyu, X.; Cheema, M.S.; Ausio, J.; Taylor, J.; Corces, V.G. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. Cell. Rep. 2017, 18, 1366–1382. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramskold, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frequency of Embryos (%) | ||||
|---|---|---|---|---|
| Timing of NEBD | ||||
| 9.0–14.5 HPI | 15.0–16.0 HPI | 16.5–17.5 HPI | 18.0+ HPI | |
| Control | 3/37 (8.1) | 8/37 (21.6) | 18/37 (48.6) | 8/37 (21.6) |
| SER | 5/43 (11.6) | 17/43 (39.5) | 16/43 (37.2) | 5/43 (11.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourzani, D.A.; Yin, Q.; Jackson, E.A.; Rando, O.J.; Visconti, P.E.; Gervasi, M.G. Sperm Energy Restriction and Recovery (SER) Alters Epigenetic Marks during the First Cell Cycle of Development in Mice. Int. J. Mol. Sci. 2023, 24, 640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010640
Tourzani DA, Yin Q, Jackson EA, Rando OJ, Visconti PE, Gervasi MG. Sperm Energy Restriction and Recovery (SER) Alters Epigenetic Marks during the First Cell Cycle of Development in Mice. International Journal of Molecular Sciences. 2023; 24(1):640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010640
Chicago/Turabian StyleTourzani, Darya A., Qiangzong Yin, Erica A. Jackson, Oliver J. Rando, Pablo E. Visconti, and Maria G. Gervasi. 2023. "Sperm Energy Restriction and Recovery (SER) Alters Epigenetic Marks during the First Cell Cycle of Development in Mice" International Journal of Molecular Sciences 24, no. 1: 640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010640