
Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Components of Protein Aggregates
3. Protein Aggregation Caused by Losing of Proteostasis
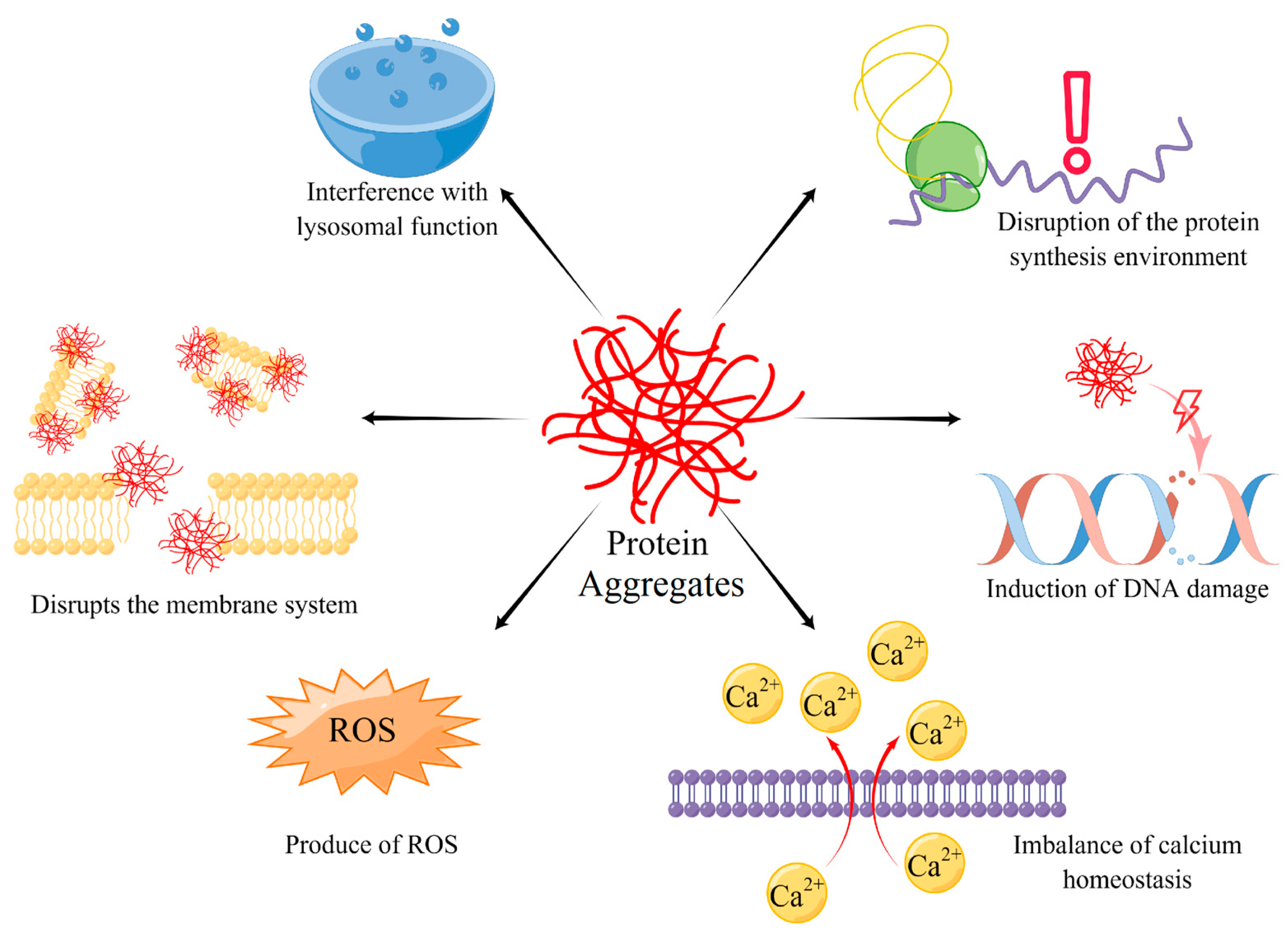
4. The Harm of Protein Aggregates
4.1. Interference with Lysosomal Function
4.2. Disruption of the Protein Synthesis Environment
4.3. Induction of DNA Damage
4.4. Imbalance of Calcium Homeostasis
4.5. Disrupts the Membrane System and Production of ROS
5. The Way to Eliminate Protein Aggregates
6. The Function of Protein Aggregates in Development of Aging-Related Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Knowles, T.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Llamas, E.; Alirzayeva, H.; Loureiro, R.; Vilchez, D. The intrinsic proteostasis network of stem cells. Curr. Opin. Cell Biol. 2020, 67, 46–55. [Google Scholar] [CrossRef]
- Bohnert, K.A.; Kenyon, C. A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature 2017, 551, 629–633. [Google Scholar] [CrossRef]
- Cheng, J.; North, B.J.; Zhang, T.; Dai, X.; Tao, K.; Guo, J.; Wei, W. The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell 2018, 17, e12801. [Google Scholar] [CrossRef]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef]
- Sanbe, A.; Osinska, H.; Saffitz, J.E.; Glabe, C.G.; Kayed, R.; Maloyan, A.; Robbins, J. Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10132–10136. [Google Scholar] [CrossRef]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef]
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. [Google Scholar] [CrossRef]
- Zaman, M.; Khan, A.N.; Zakariya, S.M.; Khan, R.H. Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: An overview and therapeutic strategies to inhibit aggregation. Int. J. Biol. Macromol. 2019, 134, 1022–1037. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [Google Scholar] [CrossRef]
- Abyzov, A.; Blackledge, M.; Zweckstetter, M. Conformational Dynamics of Intrinsically Disordered Proteins Regulate Biomolecular Condensate Chemistry. Chem. Rev. 2022, 122, 6719–6748. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Uversky, V.N. The Mysterious Unfoldome: Structureless, Underappreciated, Yet Vital Part of Any Given Proteome. J. Biomed. Biotechnol. 2009, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.P.; Kolaitis, R.-M.; Kim, H.J.; O’donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.W.; Kear-Scott, J.L.; Pilipenko, E.V.; Schwartz, M.H.; Laskowski, P.R.; Rojek, A.E.; Katanski, C.D.; Riback, J.A.; Dion, M.F.; Franks, A.M.; et al. Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress. Cell 2015, 162, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Tauber, D.; Tauber, G.; Khong, A.; Van Treeck, B.; Pelletier, J.; Parker, R. Modulation of RNA Condensation by the DEAD-Box Protein eIF4A. Cell 2020, 180, 411–426.e16. [Google Scholar] [CrossRef]
- Louka, A.; Zacco, E.; Temussi, P.A.; Tartaglia, G.G.; Pastore, A. RNA as the stone guest of protein aggregation. Nucleic Acids Res. 2020, 48, 11880–11889. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2020, 22, 183–195. [Google Scholar] [CrossRef]
- Johnson, B.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is Intrinsically Aggregation-prone, and Amyotrophic Lateral Sclerosis-linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Suram, A.; Hegde, M.; Rao, K. A new evidence for DNA nicking property of amyloid β-peptide (1–42): Relevance to Alzheimer’s disease. Arch. Biochem. Biophys. 2007, 463, 245–252. [Google Scholar] [CrossRef]
- Armstrong, R.A.; Lantos, P.L.; Cairns, N.J. What determines the molecular composition of abnormal protein aggregates in neurodegenerative disease? Neuropathology 2008, 28, 351–365. [Google Scholar] [CrossRef]
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.-H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Xu, N.; Gulick, J.; Osinska, H.; Yu, Y.; McLendon, P.M.; Shay-Winkler, K.; Robbins, J.; Yutzey, K.E. Ube2v1 Positively Regulates Protein Aggregation by Modulating Ubiquitin Proteasome System Performance Partially Through K63 Ubiquitination. Circ. Res. 2020, 126, 907–922. [Google Scholar] [CrossRef]
- Zellner, A.; Scharrer, E.; Arzberger, T.; Oka, C.; Domenga-Denier, V.; Joutel, A.; Lichtenthaler, S.F.; Müller, S.A.; Dichgans, M.; Haffner, C. CADASIL brain vessels show a HTRA1 loss-of-function profile. Acta Neuropathol. 2018, 136, 111–125. [Google Scholar] [CrossRef]
- Cheema, M.U.; Poulsen, E.T.; Enghild, J.J.; Hoorn, E.; Fenton, R.A.; Praetorius, J. Aldosterone and angiotensin II induce protein aggregation in renal proximal tubules. Physiol. Rep. 2013, 1, e00064. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread Aggregation and Neurodegenerative Diseases Are Associated with Supersaturated Proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef]
- Amzallag, E.; Hornstein, E. Crosstalk between Biomolecular Condensates and Proteostasis. Cells 2022, 11, 2415. [Google Scholar] [CrossRef]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Heaven, M.R.; Flint, D.; Randall, S.M.; Sosunov, A.A.; Wilson, L.; Barnes, S.; Goldman, J.E.; Muddiman, D.C.; Brenner, M. Composition of Rosenthal Fibers, the Protein Aggregate Hallmark of Alexander Disease. J. Proteome Res. 2016, 15, 2265–2282. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. The “Correctly Folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef]
- Zbinden, A.; Pérez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Ling, D.; Song, H.-J.; Garza, D.; Neufeld, T.P.; Salvaterra, P.M. Abeta42-Induced Neurodegeneration via an Age-Dependent Autophagic-Lysosomal Injury in Drosophila. PLoS ONE 2009, 4, e4201. [Google Scholar] [CrossRef]
- Feng, Q.; Luo, Y.; Zhang, X.-N.; Yang, X.-F.; Hong, X.-Y.; Sun, D.-S.; Li, X.-C.; Hu, Y.; Li, X.-G.; Zhang, J.-F.; et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: A vicious cycle in Alzheimer neurodegeneration. Autophagy 2019, 16, 641–658. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Jackson, G.R. The downward spiral of tau and autolysosomes: A new hypothesis in neurodegeneration. Autophagy 2012, 8, 1144–1145. [Google Scholar] [CrossRef]
- Tu, H.; Yuan, B.; Hou, X.; Zhang, X.; Pei, C.; Ma, Y.; Yang, Y.; Fan, Y.; Qin, Z.; Liu, C.; et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 2021, 110, 436–451.e11. [Google Scholar] [CrossRef]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef]
- Frakes, A.E.; Metcalf, M.G.; Tronnes, S.U.; Bar-Ziv, R.; Durieux, J.; Gildea, H.K.; Kandahari, N.; Monshietehadi, S.; Dillin, A. Four glial cells regulate ER stress resistance and longevity via neuropeptide signaling in C. elegans. Science 2020, 367, 436–440. [Google Scholar] [CrossRef]
- Schubert, D.; Behl, C.; Lesley, R.; Brack, A.; Dargusch, R.; Sagara, Y.; Kimura, H. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 1989–1993. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, A.; Huiting, W.; Barazzuol, L.; Bergink, S. Genome instability and loss of protein homeostasis: Converging paths to neurodegeneration? Open Biol. 2021, 11, 200296. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, T.R.; Kour, S.; Anderson, E.N.; Ward, C.; Rajasundaram, D.; Donnelly, C.J.; Hermann, A.; Wyne, H.; Shewmaker, F.; Pandey, U.B. DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner. Acta Neuropathol. 2021, 142, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, V.; Mitra, J.; Hegde, P.M.; Pandey, A.; Sengupta, S.; Mitra, S.; Rao, K.; Hegde, M.L. Chromatin-Bound Oxidized α-Synuclein Causes Strand Breaks in Neuronal Genomes in in vitro Models of Parkinson’s Disease. J. Alzheimer’s Dis. 2017, 60, S133–S150. [Google Scholar] [CrossRef] [PubMed]
- Weissman, L.; Jo, D.-G.; Sørensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar Amyloid Protein Aggregates Share Common Features of Cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef]
- Bode, D.C.; Freeley, M.; Nield, J.; Palma, M.; Viles, J.H. Amyloid-β oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J. Biol. Chem. 2019, 294, 7566–7572. [Google Scholar] [CrossRef]
- Valincius, G.; Heinrich, F.; Budvytyte, R.; Vanderah, D.J.; McGillivray, D.; Sokolov, Y.; Hall, J.E.; Lösche, M. Soluble Amyloid β-Oligomers Affect Dielectric Membrane Properties by Bilayer Insertion and Domain Formation: Implications for Cell Toxicity. Biophys. J. 2008, 95, 4845–4861. [Google Scholar] [CrossRef]
- Sokolov, Y.; Kozak, J.A.; Kayed, R.; Chanturiya, A.; Glabe, C.; Hall, J.E. Soluble Amyloid Oligomers Increase Bilayer Conductance by Altering Dielectric Structure. J. Gen. Physiol. 2006, 128, 637–647. [Google Scholar] [CrossRef]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of Lipid Bilayers Is a Common Conformation-dependent Activity of Soluble Amyloid Oligomers in Protein Misfolding Diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide—Insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef]
- Canale, C.; Oropesa-Nuñez, R.; Diaspro, A.; Dante, S. Amyloid and membrane complexity: The toxic interplay revealed by AFM. Semin. Cell Dev. Biol. 2017, 73, 82–94. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem. Int. Ed. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Green, J.; Kreplak, L.; Goldsbury, C.; Blatter, X.L.; Stolz, M.; Cooper, G.; Seelig, A.; Kistler, J.; Aebi, U. Atomic Force Microscopy Reveals Defects Within Mica Supported Lipid Bilayers Induced by the Amyloidogenic Human Amylin Peptide. J. Mol. Biol. 2004, 342, 877–887. [Google Scholar] [CrossRef]
- Sasahara, K.; Morigaki, K.; Shinya, K. Effects of membrane interaction and aggregation of amyloid β-peptide on lipid mobility and membrane domain structure. Phys. Chem. Chem. Phys. 2013, 15, 8929–8939. [Google Scholar] [CrossRef]
- Butterfield, D.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free. Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef]
- Singhvi, A.; Garriga, G. Asymmetric divisions, aggresomes and apoptosis. Trends Cell Biol. 2009, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, B.; Zaffagnini, G.; Fracchiolla, D.; Turco, E.; Abert, C.; Romanov, J.; Martens, S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. eLife 2015, 4, e08941. [Google Scholar] [CrossRef]
- Akopian, D.; Rape, M. Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Shah, H.V.; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol. Cell 2020, 80, 779–795.e10. [Google Scholar] [CrossRef]
- Rujano, M.A.; Bosveld, F.; Salomons, F.A.; Dijk, F.; Van Waarde, M.A.W.H.; van der Want, J.; De Vos, R.A.I.; Brunt, E.R.; Sibon, O.C.M.; Kampinga, H.H. Polarised Asymmetric Inheritance of Accumulated Protein Damage in Higher Eukaryotes. PLoS Biol. 2006, 4, e417. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Gao, Y.-S.; Sztul, E. Hassles with Taking Out the Garbage: Aggravating Aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef]
- Rebollo, E.; Sampaio, P.; Januschke, J.; Llamazares, S.; Varmark, H.; González, C. Functionally Unequal Centrosomes Drive Spindle Orientation in Asymmetrically Dividing Drosophila Neural Stem Cells. Dev. Cell 2007, 12, 467–474. [Google Scholar] [CrossRef]
- Rusan, N.M.; Peifer, M. A role for a novel centrosome cycle in asymmetric cell division. J. Cell Biol. 2007, 177, 13–20. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Mahowald, A.P.; Perlin, J.R.; Fuller, M.T. Asymmetric Inheritance of Mother Versus Daughter Centrosome in Stem Cell Division. Science 2007, 315, 518–521. [Google Scholar] [CrossRef]
- Cordes, S.; Frank, C.A.; Garriga, G. The C. elegans MELK ortholog PIG-1 regulates cell size asymmetry and daughter cell fate in asymmetric neuroblast divisions. Development 2006, 133, 2747–2756. [Google Scholar] [CrossRef]
- Frank, C.A.; Hawkins, N.C.; Guenther, C.; Horvitz, H.R.; Garriga, G.C. elegans HAM-1 positions the cleavage plane and regulates apoptosis in asymmetric neuroblast divisions. Dev. Biol. 2005, 284, 301–310. [Google Scholar] [CrossRef]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef]
- Natalello, A.; Liu, J.; Ami, D.; Doglia, S.M.; de Marco, A. The osmolyte betaine promotes protein misfolding and disruption of protein aggregates. Proteins Struct. Funct. Bioinform. 2008, 75, 509–517. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Park, C.B.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M.M. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines 2022, 10, 3276. [Google Scholar] [CrossRef]
- Hirohata, M.; Ono, K.; Naiki, H.; Yamada, M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology 2005, 49, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Andrich, K.; Bieschke, J. The Effect of (−)-Epigallo-catechin-(3)-gallate on Amyloidogenic Proteins Suggests a Common Mechanism. Nat. Compd. Ther. Agents Amyloidogenic Dis. 2015, 863, 139–161. [Google Scholar] [CrossRef]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef]
- McGrail, D.J.; Garnett, J.; Yin, J.; Dai, H.; Shih, D.J.; Lam, T.N.A.; Li, Y.; Sun, C.; Li, Y.; Schmandt, R.; et al. Proteome Instability Is a Therapeutic Vulnerability in Mismatch Repair-Deficient Cancer. Cancer Cell 2020, 37, 371–386.e12. [Google Scholar] [CrossRef]
- Kumar, S.; Stokes, J.; Singh, U.P.; Gunn, K.S.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.-H.; He, X.-H.; Feng, Z.-S.; Li, D.-Y.; Tang, J.-X.; Liu, H.-F. Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination. Int. J. Mol. Sci. 2023, 24, 8593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108593
Wen J-H, He X-H, Feng Z-S, Li D-Y, Tang J-X, Liu H-F. Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination. International Journal of Molecular Sciences. 2023; 24(10):8593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108593
Chicago/Turabian StyleWen, Jun-Hao, Xiang-Hong He, Ze-Sen Feng, Dong-Yi Li, Ji-Xin Tang, and Hua-Feng Liu. 2023. "Cellular Protein Aggregates: Formation, Biological Effects, and Ways of Elimination" International Journal of Molecular Sciences 24, no. 10: 8593. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108593