
Novel Polymyxin-Inspired Peptidomimetics Targeting the SARS-CoV-2 Spike:hACE2 Interface

 , , , , , , , , , and
, , , , , , , , , and
Abstract
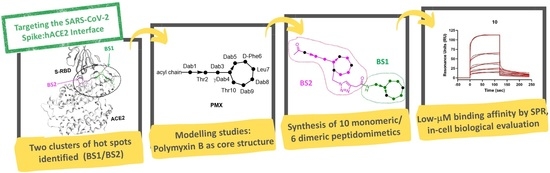
:
1. Introduction

2. Results
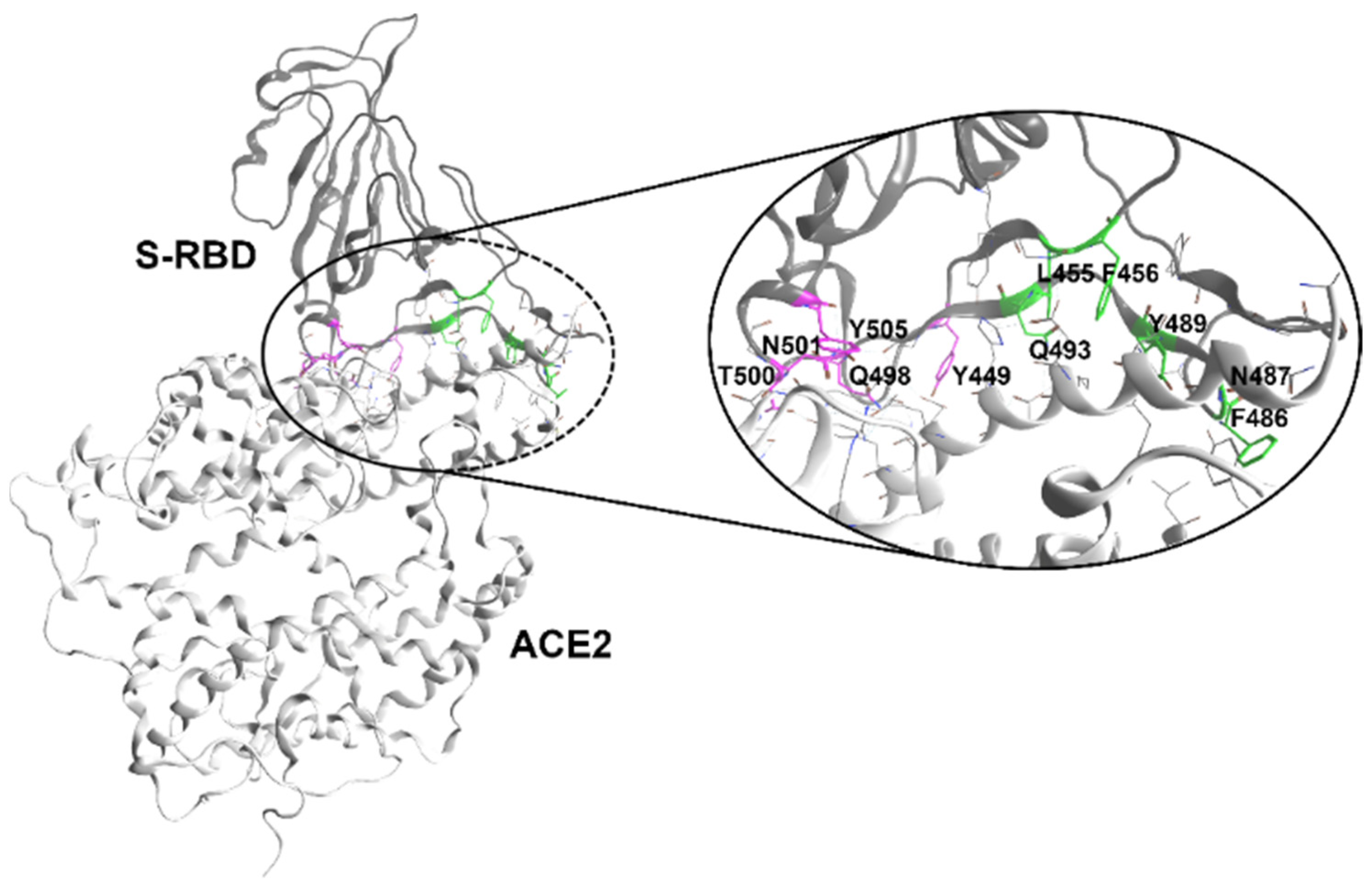
2.1. In Silico Design of Polymyxin B-Based Peptidomimetics
2.2. Chemical Synthesis of Polymyxin-Based Peptidomimetics
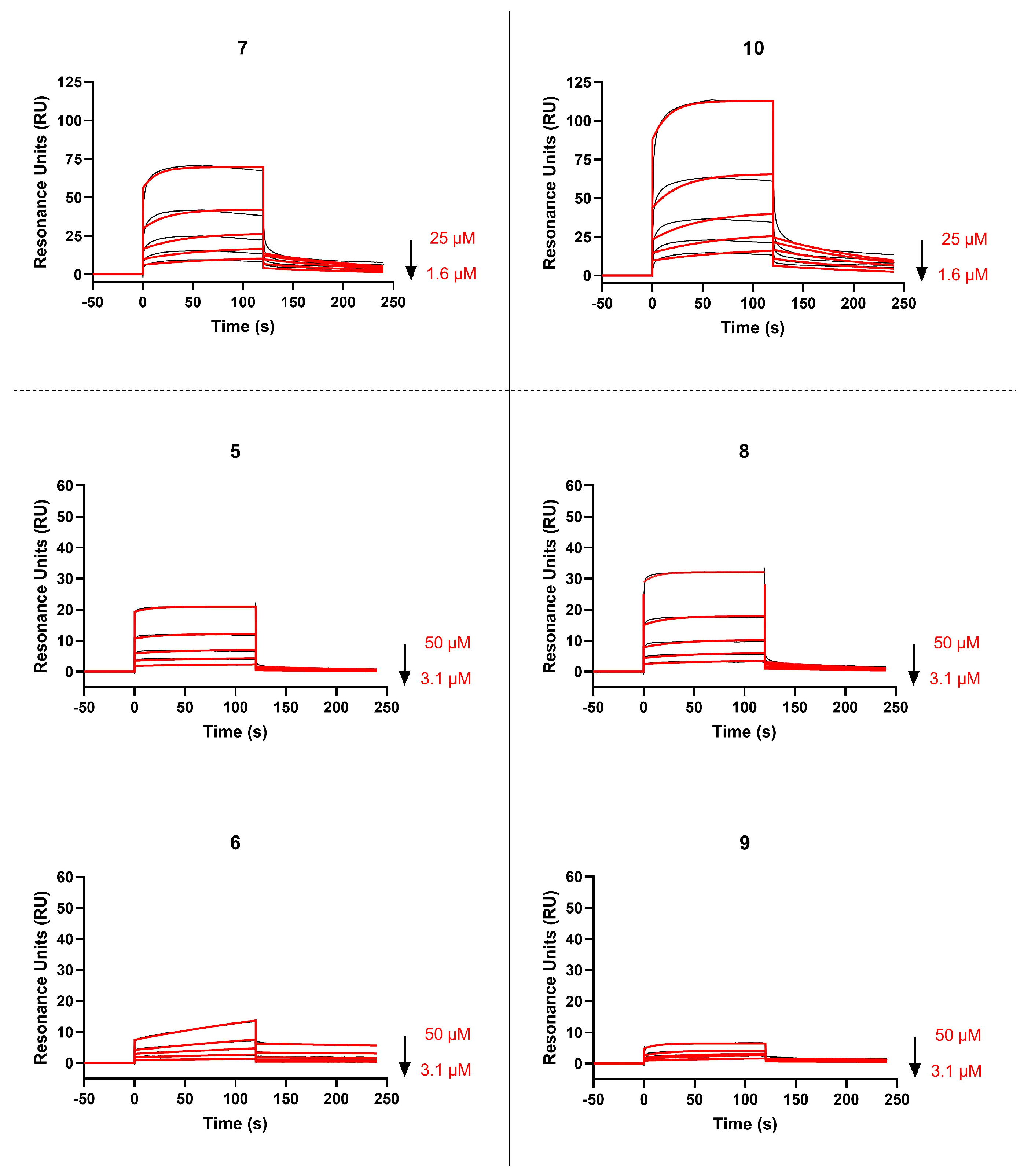
2.3. Binding Affinity of Polymyxin-Based Peptidomimetics to Spike RBD by SPR Analysis
2.4. Antiviral Effect of PMX-Inspired Peptidomimetics against SARS-CoV-2
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling
4.2. Chemistry
4.2.1. General Procedure A to BS1-Targeted Monomers as Exemplified by the Synthesis of Monomer 1
4.2.2. General Procedure B to BS2-Targeted Monomers as Exemplified by the Synthesis of Monomer 2
4.2.3. General Procedure C to BS1/BS2-Targeted Heterodimers as Exemplified by the Synthesis of Dimer 3 (Scheme 3)
4.3. Biology
4.3.1. Surface Plasmon Resonance
4.3.2. Wild-Type Virus Infection and Antiviral Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- European Centre for Disease Prevention and Control. 2023. Available online: https://www.ecdc.europa.eu/en/covid-19/situation-updates (accessed on 19 February 2023).
- Mathieu, E.; Ritchie, H.; Rodés-Guirao, L.; Appel, C.; Gavrilov, D.; Giattino, C.; Hasell, J.; Macdonald, B.; Dattani, S.; Beltekian, D.; et al. Coronavirus Pandemic (COVID-19). 2023. Available online: https://ourworldindata.org/coronavirus (accessed on 19 February 2023).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- NIH. COVID-19 Treatment Guidelines. 2023. Available online: https://www.covid19treatmentguidelines.nih.gov/about-the-guidelines/whats-new/ (accessed on 20 February 2023).
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Mehra, R.; Keep, K.P. Structural heterogeneity and precision of implications drawn from cryo-electron microscopy structures: SARS-CoV-2 spike-protein mutations as a test case. ACS Infect. Dis. 2022, 8, 29–58. [Google Scholar] [CrossRef]
- Moccia, F.; Gerbino, A.; Lionetti, V.; Miragoli, M.; Munaron, L.M.; Pagliaro, P.; Pasqua, T.; Penna, C.; Rocca, C.; Samaja, M.; et al. COVID-19-associated cardiovascular morbidity in older adults: A position paper from the Italian Society of Cardiovascular Researches. GeroScience 2020, 42, 1021–1049. [Google Scholar] [CrossRef]
- Razeghian-Jahromi, I.; Zibaeenezhad, M.J.; Lu, Z.; Zahra, E.; Mahboobeh, R.; Lionetti, V. Angiotensin-converting enzyme 2: A double-edged sword in COVID-19 patients with an increased risk of heart failure. Heart Fail. Rev. 2021, 26, 371–380. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef]
- Del Rio, C.; Collins, L.F.; Malani, P. Long-term Health Consequences of COVID-19. JAMA 2020, 324, 1723–1724. [Google Scholar] [CrossRef]
- Chitsike, L.; Duerksen-Hughes, P. Keep out! SARS-CoV-2 entry inhibitors: Their role and utility as COVID-19 therapeutics. Virol. J. 2021, 18, 154. [Google Scholar] [CrossRef]
- Krumm, Z.A.; Lloyd, G.M.; Francis, C.P.; Nasif, L.H.; Mitchell, D.A.; Golde, T.E.; Giasson, B.I.; Xia, Y. Precision therapeutic targets for COVID-19. Virol. J. 2021, 18, 66. [Google Scholar] [CrossRef]
- Twomey, J.D.; Luo, S.; Dean, A.Q.; Bozza, W.P.; Nalli, A.; Zhang, B. COVID-19 update: The race to therapeutic development. Drug Resist. Updates 2020, 53, 100733. [Google Scholar] [CrossRef]
- Zhou, T.; Tsybovsky, Y.; Olia, A.S.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.Y.; Katsamba, P.S.; Nazzari, A.; Sampson, J.M.; et al. Cryo-EM Structures Delineate a pH-Dependent Switch that Mediates Endosomal Positioning of SARS-CoV-2 Spike Receptor-Binding Domains. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Melby, T.; Westby, M. Inhibitors of viral entry. Handb. Exp. Pharmacol. 2009, 189, 177–202. [Google Scholar]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Lv, Z.; Deng, Y.Q.; Ye, Q.; Cao, L.; Sun, C.Y.; Fan, C.; Huang, W.; Sun, S.; Sun, Y.; Zhu, L.; et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science 2020, 369, 1505–1509. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Jia, H.; Neptune, E.; Cui, H. Targeting ACE2 for COVID-19 Therapy: Opportunities and Challenges. Am. J. Respir. Cell Mol. Biol. 2021, 64, 416–425. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Pomplun, S. Targeting the SARS-CoV-2-spike protein: From antibodies to miniproteins and peptides. RSC Med. Chem. 2021, 12, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Ferreira, L.; Hwang, P.; Xu, J.; Stroud, R. Peptide Antidotes to SARS-CoV-2 (COVID-19). bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Loas, A.; Pentelute, B.L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. bioRxiv 2020. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.K.; Hillyer, C.D.; Debnath, A.K. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection In Vitro. MBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Morgan, D.C.; Morris, C.; Mahindra, A.; Blair, C.M.; Tejeda, G.; Herbert, I.; Turnbull, M.L.; Lieber, G.; Willett, B.J.; Logan, N.; et al. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Pept. Sci. 2021, 113, e24217. [Google Scholar] [CrossRef]
- Karoyan, P.; Vieillard, V.; Gomez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.E.; Denis, A.; Grondin, P.; Lequin, O. A Collection of Designed Peptides to Target SARS-CoV-2 Spike RBD—ACE2 Interaction. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef]
- Han, S.; Zhao, G.; Wei, Z.; Chen, Y.; Zhao, J.; He, Y.; He, Y.J.; Gao, J.; Chen, S.; Du, C.; et al. An angiotensin-converting enzyme-2-derived heptapeptide GK-7 for SARS-CoV-2 spike blockade. Peptides 2021, 145, 170638. [Google Scholar] [CrossRef]
- Sadremomtaz, A.; Al-Dahmani, Z.M.; Ruiz-Moreno, A.J.; Monti, A.; Wang, C.; Azad, T.; Bell, J.C.; Doti, N.; Velasco-Velázquez, M.A.; de Jong, D.; et al. Synthetic Peptides That Antagonize the Angiotensin-Converting Enzyme-2 (ACE-2) Interaction with SARS-CoV-2 Receptor Binding Spike Protein. J. Med. Chem. 2022, 65, 2836–2847. [Google Scholar] [CrossRef]
- Larue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.K.; Sharma, A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconj. Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef]
- Souza, P.F.N.; Marques, L.S.M.; Oliveira, J.T.A.; Lima, P.G.; Dias, L.P.; Neto, N.A.S.; Lopes, F.E.S.; Sousa, J.S.; Silva, A.F.B.; Caneiro, R.F.; et al. Synthetic antimicrobial peptides: From choice of the best sequences to action mechanisms. Biochimie 2020, 175, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Calugi, L.; Sautariello, G.; Lenci, E.; Mattei, M.L.; Coppa, C.; Cini, N.; Contini, A.; Trabocchi, A. Identification of a short ACE2-derived stapled peptide targeting the SARS-CoV-2 spike protein. Eur. J. Med. Chem. 2023, 249, 115118. [Google Scholar] [CrossRef] [PubMed]
- Though classification of peptides based on their length is not stringent, in this work the term “medium-sized” refers to peptide mimics corresponding to 10-to-30-mer peptides.
- Nguyen, H.; Lan, P.D.; Nissley, D.A.; O’Brien, E.P.; Li, M.S. Cocktail of REGN Antibodies Binds More Strongly to SARS-CoV-2 Than Its Components, but the Omicron Variant Reduces Its Neutralizing Ability. J. Phys. Chem. B 2022, 126, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Wade, J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014, 2, 62. [Google Scholar] [CrossRef]
- Rubin, S.; Qvit, N. Cyclic Peptides for Protein-Protein Interaction Targets: Applications to Human Disease. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 199–221. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. In Silico Drug Repurposing for SARS-CoV-2 Main Proteinase and Spike Proteins. J. Proteome Res. 2020, 19, 4637–4648. [Google Scholar] [CrossRef]
- Storm, D.R.; Rosenthal, K.S.; Swanson, P.E. Polymyxin and related peptide antibiotics. Annu. Rev. Biochem. 1977, 46, 723–763. [Google Scholar] [CrossRef]
- Kassamali, Z.; Rotschafer, J.C.; Jones, R.N.; Prince, R.A.; Danziger, L.H. Polymyxins: Wisdom does not always come with age. Clin. Infect. Dis. 2013, 57, 877–883. [Google Scholar] [CrossRef]
- Segovia, R.; Solé, J.; Marqués, A.M.; Cajal, Y.; Rabanal, F. Unveiling the Membrane and Cell Wall Action of Antimicrobial Cyclic Lipopeptides: Modulation of the Spectrum of Activity. Pharmaceutics 2021, 13, 2180. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, B.K. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Harris, P.W.R.; Brimble, M.A. On-Resin Preparation of Allenamidyl Peptides: A Versatile Chemoselective Conjugation and Intramolecular Cyclisation Tool. Angew. Chem. Int. Ed. 2020, 59, 18054–18061. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.L.; Cui, A.L.; Hu, X.X.; You, X.F.; Li, Z.R.; Zheng, J.S. A new strategy for total solid-phase synthesis of polymyxins. Tetrahedron Lett. 2015, 56, 4796–4799. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Chan, W.C.; Chhabra, S.R.; Hone, N.D. A novel lysine-protecting procedure for continuous flow solid phase synthesis of branched peptides. J. Chem. Soc. Chem. Commun. 1993, 9, 778–779. [Google Scholar] [CrossRef]
- Vanhulle, E.; D’huys, T.; Provinciael, B.; Stroobants, J.; Camps, A.; Noppen, S.; Schols, D.; Van Damme, E.J.M.; Maes, P.; Stevaert, A.; et al. Carbohydrate-binding protein from stinging nettle as fusion inhibitor for SARS-CoV-2 variants of concern. Front. Cell. Infect. Microbiol. 2022, 12, 989534. [Google Scholar] [CrossRef] [PubMed]
- Vanhulle, E.; Stroobants, J.; Provinciael, B.; Camps, A.; Noppen, S.; Maes, P.; Vermeire, K. SARS-CoV-2 Permissive glioblastoma cell line for high throughput antiviral screening. Antivir. Res. 2022, 203, 105342. [Google Scholar] [CrossRef]
- With these marked deviations from the original PMX structure, it is presumable that the PMs of this work are deprived of the antibacterial activity of PMX. Biological evaluation of antibacterial activity of PMs was not carried out here, going beyond the scope of this antiviral work.
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2020.
- Labute, P. LowModeMD—Implicit Low-Mode Velocity Filtering Applied to Conformational Search of Macrocycles and Protein Loops. J. Chem. Inf. Model. 2010, 50, 792–800. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanskia, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Pellegrino, S.; Clayden, J.; Contini, A. Mechanism of Stabilization of Helix Secondary Structure by Constrained Cα-Tetrasubstituted α-Amino Acids. J. Phys. Chem. B 2015, 119, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, I.; Clayden, J.; Contini, A. Origin of Helical Screw Sense Selectivity Induced by Chiral Constrained Cα-Tetrasubstituted α-Amino Acids in Aib-based Peptides. J. Phys. Chem. B 2015, 119, 14003–14013. [Google Scholar] [CrossRef] [PubMed]
- Tomsett, M.; Maffucci, I.; Le Bailly, B.A.F.; Byrne, L.; Bijvoets, S.M.; Lizio, M.G.; Raftery, J.; Butts, C.P.; Webb, S.J.; Contini, A.; et al. A tendril perversion in a helical oligomer: Trapping and characterizing a mobile screw-sense reversal. J. Chem. Sci. 2017, 8, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Vassetti, D.; Pagliai, M.; Procacci, P. Assessment of GAFF2 and OPLS-AA General Force Fields in Combination with the Water Models TIP3P, SPCE, and OPC3 for the Solvation Free Energy of Druglike Organic Molecules. J. Chem. Theory Comput. 2019, 15, 1983–1995. [Google Scholar] [CrossRef]
- Prabhudesai, K.S.; Raje, S.; Dhamanaskar, A.; Modi, D.; Dighe, V.; Contini, A.; Idicula-Thomas, S. Central residues of FSHβ (89-97) peptide are not critical for FSHR binding: Implications for peptidomimetic design. Peptides 2020, 132, 170367. [Google Scholar] [CrossRef]
- Macut, H.; Hu, X.; Tarantino, D.; Gilardoni, E.; Clerici, F.; Regazzoni, L.; Contini, A.; Pellegrino, S.; Gelmi, M.L. Tuning PFKFB3 Bisphosphatase Activity Through Allosteric Interference. Sci. Rep. 2019, 9, 20333. [Google Scholar] [CrossRef]
- Giatti, S.; Di Domizio, A.; Diviccaro, S.; Falvo, E.; Caruso, D.; Contini, A.; Melcangi, R.C. Three-Dimensional Proteome-Wide Scale Screening for the 5-Alpha Reductase Inhibitor Finasteride: Identification of a Novel Off-Target. J. Med. Chem. 2021, 64, 4553–4566. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; Darden, T.A.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Vanhulle, E.; Provinciael, B.; Stroobants, J.; Camps, A.; Maes, P.; Vermeire, K. Intracellular flow cytometry complements RT-qPCR detection of circulating SARS-CoV-2 variants of concern. BioTechniques 2022, 72, 245–254. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
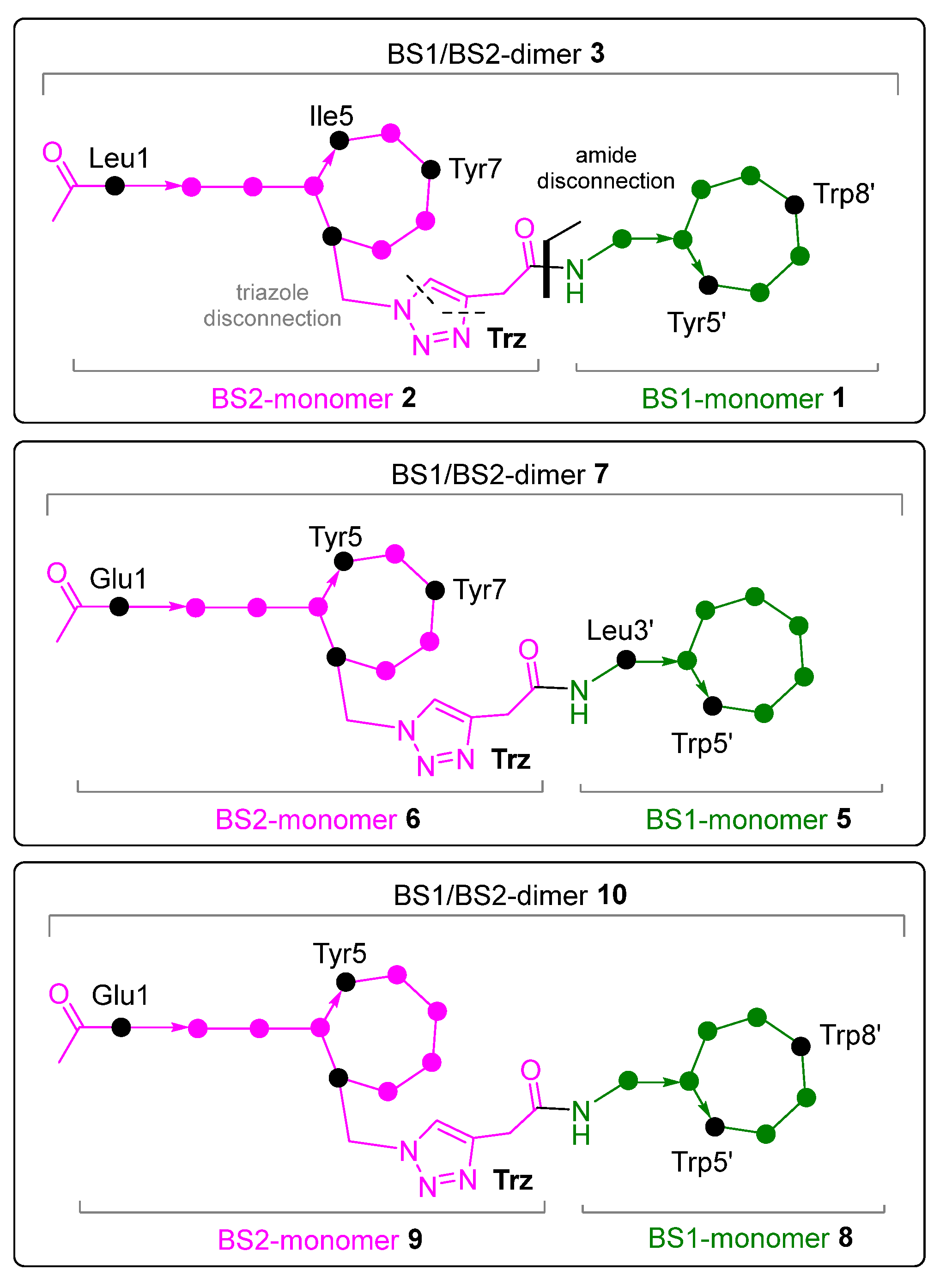{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Sequence | m/z (b) Found (calcd) | MW (Da) |
|---|---|---|---|
| PMX |  | nd | 1203.5 |
| 3 |  | 568.3075 (568.3067) [M+4H]4+ | 2269.3 |
| 4 |  | 571.5606 (571.5607) [M+4H]4+ | 2283.7 |
| 7 |  | 572.2990 (572.2964) [M+4H]4+ | 2285.6 |
| 10 |  | 578.0528 (578.0504) [M+4H]4+ | 2308.6 |
| 14 |  | 758.0576 (758.0570) [M+3H]3+ | 2272.6 |
| 16 |  | 1137.0737 (1137.0737) [M+2H]2+ | 2273.6 |
| Ligand/Analyte | ka (×103 M−1 s−1) | kd (×10−3 s−1) | KD (µM) |
|---|---|---|---|
| RBD/dimer 7 | 3.09 ± 0.59 | 6.83 ± 1.58 | 2.31 ± 0.95 |
| RBD/monomer 5 | n.a. | n.a. | n.a. |
| RBD/monomer 6 | n.a. | n.a. | n.a. |
| RBD/dimer 10 | 2.65 ± 0.67 | 7.00 ± 1.10 | 2.78 ± 1.12 |
| RBD/monomer 8 | 1.20 ± 0.41 | 9.43 ± 1.52 | 8.56 ± 4.16 |
| RBD/monomer 9 | n.a. | n.a. | n.a. |
| RBD/dimer 3 | n.a. | n.a. | n.a. |
| RBD/dimer 4 | n.a. | n.a. | n.a. |
| RBD/monomer 1 | 0.61 ± 0.05 | 6.12 ± 0.79 | 10.12 ± 2.09 |
| RBD/monomer 2 | 0.46 ± 0.02 | 4.39 ± 0.39 | 9.62 ± 1.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bugatti, K.; Sartori, A.; Battistini, L.; Coppa, C.; Vanhulle, E.; Noppen, S.; Provinciael, B.; Naesens, L.; Stevaert, A.; Contini, A.; et al. Novel Polymyxin-Inspired Peptidomimetics Targeting the SARS-CoV-2 Spike:hACE2 Interface. Int. J. Mol. Sci. 2023, 24, 8765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108765
Bugatti K, Sartori A, Battistini L, Coppa C, Vanhulle E, Noppen S, Provinciael B, Naesens L, Stevaert A, Contini A, et al. Novel Polymyxin-Inspired Peptidomimetics Targeting the SARS-CoV-2 Spike:hACE2 Interface. International Journal of Molecular Sciences. 2023; 24(10):8765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108765
Chicago/Turabian StyleBugatti, Kelly, Andrea Sartori, Lucia Battistini, Crescenzo Coppa, Emiel Vanhulle, Sam Noppen, Becky Provinciael, Lieve Naesens, Annelies Stevaert, Alessandro Contini, and et al. 2023. "Novel Polymyxin-Inspired Peptidomimetics Targeting the SARS-CoV-2 Spike:hACE2 Interface" International Journal of Molecular Sciences 24, no. 10: 8765. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108765