

Regulation of Acetylation States by Nutrients in the Inhibition of Vascular Inflammation and Atherosclerosis

{kind=link}
{kind=link}
Abstract
:1. Introduction
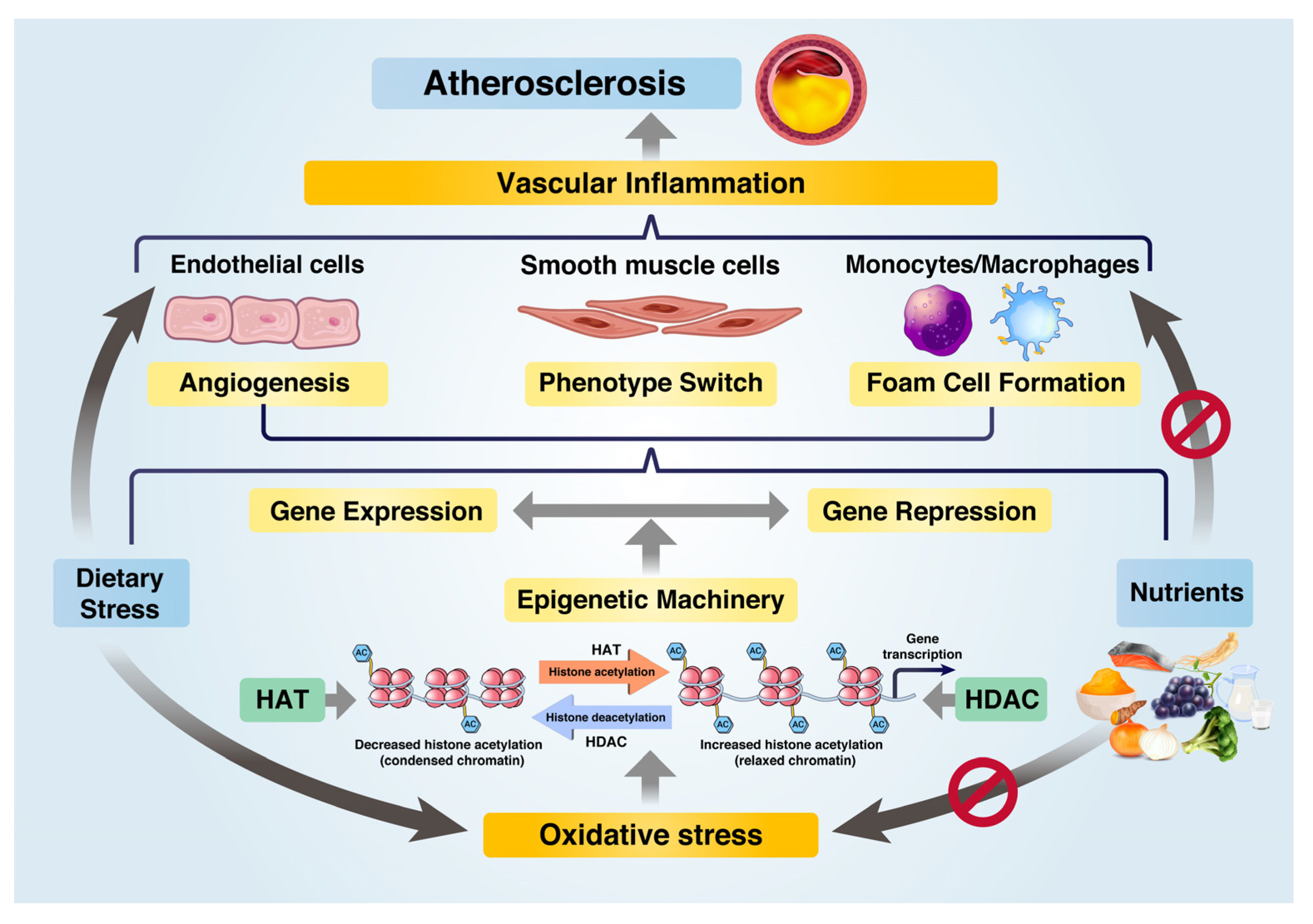
2. Epigenetic Regulation of VI and AS
2.1. Factors Affecting VI and AS
2.2. Regulation of VI and AS
3. Roles of Nutrients to Inhibit VI and AS
3.1. Roles of Nutrients in Regulating Inflammatory Responses
3.2. Roles of Nutrients in Regulating Histone Acetylation State
4. Epigenetic Roles of Nutrients in Regulating VI and AS
4.1. Astaxanthin (ASTX)
4.2. Butyrate
4.3. Curcumin
4.4. Ginsenoside
4.5. Nicotinamide Riboside (NR)
4.6. Quercetin
4.7. Resveratrol (RES)
4.8. Sulforaphane (SFN)
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective Effects of Sirtuins in Cardiovascular Diseases: From Bench to Bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.J.; Stroes, E.S.G.; Bahjat, M. Potential Epigenetic Therapeutics for Atherosclerosis Treatment. Atherosclerosis 2019, 281, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox Regulation of SIRT1 in Inflammation and Cellular Senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Neele, A.E.; Hoeksema, M.A.; de Heij, F.; Boshuizen, M.C.; van der Velden, S.; de Boer, V.C.; Reedquist, K.A.; de Winther, M.P. Inhibiting Epigenetic Enzymes to Improve Atherogenic Macrophage Functions. Biochem. Biophys. Res. Commun. 2014, 455, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Lawn, R.M. ABCA1. The Gatekeeper for Eliminating Excess Tissue Cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 Regulates Hepatocyte Lipid Metabolism through Activating AMP-Activated Protein Kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.; Netea, M.G.; Gomes, M.E.; et al. Innate Immune Cell Activation and Epigenetic Remodeling in Symptomatic and Asymptomatic Atherosclerosis in Humans in Vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 2011, 10, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting Epigenetics and Non-Coding RNAs in Atherosclerosis: From Mechanisms to Therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef]
- Yao, B.C.; Meng, L.B.; Hao, M.L.; Zhang, Y.M.; Gong, T.; Guo, Z.G. Chronic Stress: A Critical Risk Factor for Atherosclerosis. J. Int. Med. Res. 2019, 47, 1429–1440. [Google Scholar] [CrossRef]
- Marin, T.; Gongol, B.; Chen, Z.; Woo, B.; Subramaniam, S.; Chien, S.; Shyy, J.Y.J. Mechanosensitive MicroRNAs—Role in Endothelial Responses to Shear Stress and Redox State. Free Radic. Biol. Med. 2013, 64, 61–68. [Google Scholar] [CrossRef]
- Libby, P. History of Discovery: Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; Anderson, K.R.; Topper, J.N. The Critical Role of Mechanical Forces in Blood Vessel Development, Physiology and Pathology. J. Vasc. Surg. 1999, 29, 1104–1151. [Google Scholar] [CrossRef]
- Nigro, P.; Abe, J.I.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414. [Google Scholar] [CrossRef]
- Pandey, D.; Hori, D.; Kim, J.H.; Bergman, Y.; Berkowitz, D.E.; Romer, L.H. NEDDylation Promotes Endothelial Dysfunction: A Role for HDAC2. J. Mol. Cell. Cardiol. 2015, 81, 18–22. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.X.; Zhou, T.; Wang, X.A.; Tong, X.H.; Ding, J.W. Histone Deacetylases and Atherosclerosis. Atherosclerosis 2015, 240, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; Van den Bossche, J.; Hoeksema, M.A.; de Winther, M.P. Epigenetic Pathways in Macrophages Emerge as Novel Targets in Atherosclerosis. Eur. J. Pharmacol. 2015, 763, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P. The Immune Response in Atherosclerosis: A Double-Edged Sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Doyle, B.; Caplice, N. Plaque Neovascularization and Antiangiogenic Therapy for Atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 2073–2080. [Google Scholar] [CrossRef]
- Bae, J.U.; Lee, S.J.; Seo, K.W.; Kim, Y.H.; Park, S.Y.; Bae, S.S.; Kim, C.D. SIRT1 Attenuates Neointima Formation by Inhibiting HIF-1alpha Expression in Neointimal Lesion of a Murine Wire-Injured Femoral Artery. Int. J. Cardiol. 2013, 168, 4393–4396. [Google Scholar] [CrossRef]
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, X.; Long, C.Y.; Pi, Y.; Gao, C.Y.; Li, J.C.; Zhang, L.L. SIRT1 Improves VSMC Functions in Atherosclerosis. Prog. Biophys. Mol. Biol. 2016, 121, 11–15. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Andreeva, E.R.; Krushinsky, A.V.; Novikov, I.D.; Tertov, V.V.; Nestaiko, G.V.; Khashimov, K.A.; Repin, V.S.; Smirnov, V.N. Intimal Cells and Atherosclerosis. Relationship between the Number of Intimal Cells and Major Manifestations of Atherosclerosis in the Human Aorta. Am. J. Pathol. 1986, 125, 402–415. [Google Scholar]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Winnik, S.; Stein, S.; Matter, C.M. SIRT1—An Anti-Inflammatory Pathway at the Crossroads between Metabolic Disease and Atherosclerosis. Curr. Vasc. Pharmacol. 2012, 10, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Lohmann, C.; Schafer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Boren, J.; et al. SIRT1 Decreases Lox-1-Mediated Foam Cell Formation in Atherogenesis. Eur. Heart J. 2010, 31, 2301–2309. [Google Scholar] [CrossRef]
- Zeng, H.T.; Fu, Y.C.; Yu, W.; Lin, J.M.; Zhou, L.; Liu, L.; Wang, W. SIRT1 Prevents Atherosclerosis via Liver-X-receptor and NF-kappaB Signaling in a U937 Cell Model. Mol. Med. Rep. 2013, 8, 23–28. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A Role for the NAD-Dependent Deacetylase Sirt1 in the Regulation of Autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Li, B.-H.; Yin, Y.-W.; Liu, Y.; Pi, Y.; Guo, L.; Cao, X.-J.; Gao, C.-Y.; Zhang, L.-L.; Li, J.-C. TRPV1 Activation Impedes Foam Cell Formation by Inducing Autophagy in OxLDL-Treated Vascular Smooth Muscle Cells. Cell Death Dis. 2014, 5, e1182. [Google Scholar] [CrossRef]
- Jovinge, S.; Crisby, M.; Thyberg, J.; Nilsson, J. DNA Fragmentation and Ultrastructural Changes of Degenerating Cells in Atherosclerotic Lesions and Smooth Muscle Cells Exposed to Oxidized LDL in Vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2225–2231. [Google Scholar] [CrossRef]
- Mallat, Z.; Tedgui, A. Apoptosis in the Vasculature: Mechanisms and Functional Importance. Br. J. Pharmacol. 2000, 130, 947–962. [Google Scholar] [CrossRef]
- He, J.; Liu, X.; Su, C.; Wu, F.; Sun, J.; Zhang, J.; Yang, X.; Zhang, C.; Zhou, Z.; Zhang, X.; et al. Inhibition of Mitochondrial Oxidative Damage Improves Reendothelialization Capacity of Endothelial Progenitor Cells via SIRT3 (Sirtuin 3)-Enhanced SOD2 (Superoxide Dismutase 2) Deacetylation in Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1682–1698. [Google Scholar] [CrossRef]
- Jing, S.-H.; Yu, B.; Qiao, H. Correlation between Endothelial Cell Apoptosis and SIRT3 Gene Expression in Atherosclerosis Rats. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9033–9040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, J.; Shen, S.; Tong, Q.; Ma, X.; Lin, L. SIRT3 Promotes Lipophagy and Chaperon-Mediated Autophagy to Protect Hepatocytes against Lipotoxicity. Cell Death Differ. 2020, 27, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 Regulates Mitochondrial Fatty-Acid Oxidation by Reversible Enzyme Deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Ross, R.; Després, J.-P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and Ectopic Fat, Atherosclerosis, and Cardiometabolic Disease: A Position Statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef]
- Sheng, S.; Kang, Y.; Guo, Y.; Pu, Q.; Cai, M.; Tu, Z. Overexpression of Sirt3 Inhibits Lipid Accumulation in Macrophages through Mitochondrial IDH2 Deacetylation. Int. J. Clin. Exp. Pathol. 2015, 8, 9196–9201. [Google Scholar]
- Liu, Y.; Shen, X.; Pang, M.; Sun, Z.; Qian, Y.; Xue, W.; Wang, Z.; Li, L. Role of Histone Deacetylase Sirt3 in the Development and Regression of Atherosclerosis. Life Sci. 2021, 272, 119178. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Chen, J.X. Emerging Role of SIRT3 in Endothelial Metabolism, Angiogenesis, and Cardiovascular Disease. J. Cell. Physiol. 2019, 234, 2252–2265. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Chen, S.T.; Roman, R.J.; Aschner, J.L.; Didion, S.; Chen, J.X. Endothelial Specific SIRT3 Deletion Impairs Glycolysis and Angiogenesis and Causes Diastolic Dysfunction. J. Mol. Cell. Cardiol. 2017, 112, 104–113. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kang, H.; Kim, B. Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State. Foods 2023, 12, 925. [Google Scholar] [CrossRef] [PubMed]
- Sosnowska, B.; Mazidi, M.; Penson, P.; Gluba-Brzózka, A.; Rysz, J.; Banach, M. The Sirtuin Family Members SIRT1, SIRT3 and SIRT6: Their Role in Vascular Biology and Atherogenesis. Atherosclerosis 2017, 265, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.S.; Kim, J.M.; Kim, K.L.; Jang, S.Y.; Jeon, E.S.; Choi, S.H.; Kim, D.K.; Suh, W.; Kim, Y.W. Early Growth Response Factor-1 Is Associated with Intraluminal Thrombus Formation in Human Abdominal Aortic Aneurysm. J. Am. Coll. Cardiol. 2009, 53, 792–799. [Google Scholar] [CrossRef]
- Wang, B.; Chen, J.; Santiago, F.S.; Janes, M.; Kavurma, M.M.; Chong, B.H.; Pimanda, J.E.; Khachigian, L.M. Phosphorylation and Acetylation of Histone H3 and Autoregulation by Early Growth Response 1 Mediate Interleukin 1beta Induction of Early Growth Response 1 Transcription. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Haka, A.S.; Asmal, A.; Barbosa-Lorenzi, V.C.; Grosheva, I.; Chin, H.F.; Xiong, Y.; Hla, T.; Maxfield, F.R. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and P38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Howell, K.W.; Meng, X.; Fullerton, D.A.; Jin, C.; Reece, T.B.; Cleveland, J.C., Jr. Toll-like Receptor 4 Mediates Oxidized LDL-Induced Macrophage Differentiation to Foam Cells. J. Surg. Res. 2011, 171, e27–e31. [Google Scholar] [CrossRef]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage Plasticity in Experimental Atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef]
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Nassar, T.; Sachais, B.S.; Akkawi, S.; Kowalska, M.A.; Bdeir, K.; Leitersdorf, E.; Hiss, E.; Ziporen, L.; Aviram, M.; Cines, D.; et al. Platelet Factor 4 Enhances the Binding of Oxidized Low-Density Lipoprotein to Vascular Wall Cells. J. Biol. Chem. 2003, 278, 6187–6193. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Pang, J.; Zhang, H.; Luo, J.; Qian, X.; Chen, Q.; Ling, W. Attenuation of Atherosclerosis by Protocatechuic Acid via Inhibition of M1 and Promotion of M2 Macrophage Polarization. J. Agric. Food Chem. 2019, 67, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lee, Y.; Bae, M.; Park, Y.K.; Lee, J.Y. Astaxanthin Inhibits Alcohol-Induced Inflammation and Oxidative Stress in Macrophages in a Sirtuin 1-Dependent Manner. J. Nutr. Biochem. 2020, 85, 108477. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, Y.K.; Lee, J.Y. Inhibition of Alcohol-Induced Inflammation and Oxidative Stress by Astaxanthin Is Mediated by Its Opposite Actions in the Regulation of Sirtuin 1 and Histone Deacetylase 4 in Macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158838. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef]
- Kang, H.; Lee, Y.; Kim, M.-B.; Hu, S.; Jang, H.; Park, Y.-K.; Lee, J.-Y. The Loss of Histone Deacetylase 4 in Macrophages Exacerbates Hepatic and Adipose Tissue Inflammation in Male but Not in Female Mice with Diet-Induced Non-Alcoholic Steatohepatitis. J. Pathol. 2021, 255, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shu, L.; Huang, W.; Song, G.; Ma, H. Resveratrol Affects Hepatic Gluconeogenesis via Histone Deacetylase 4. Diabetes Metab. Syndr. Obes. 2019, 12, 401–411. [Google Scholar] [CrossRef]
- Li, M.; Hong, W.; Hao, C.; Li, L.; Xu, H.; Li, P.; Xu, Y. Hepatic Stellate Cell-Specific Deletion of SIRT1 Exacerbates Liver Fibrosis in Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3202–3211. [Google Scholar] [CrossRef]
- Fang, M.; Fan, Z.; Tian, W.; Zhao, Y.; Li, P.; Xu, H.; Zhou, B.; Zhang, L.; Wu, X.; Xu, Y. HDAC4 Mediates IFN-Gamma Induced Disruption of Energy Expenditure-Related Gene Expression by Repressing SIRT1 Transcription in Skeletal Muscle Cells. Biochim. Biophys. Acta 2016, 1859, 294–305. [Google Scholar] [CrossRef]
- He, W.; Li, Q.; Li, X. Acetyl-CoA Regulates Lipid Metabolism and Histone Acetylation Modification in Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188837. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of Pyruvate Metabolism and Human Disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide Riboside, an NAD(+) Precursor, Attenuates Inflammation and Oxidative Stress by Activating Sirtuin 1 in Alcohol-Stimulated Macrophages. Lab. Investig. 2021, 101, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, S.; Li, N.; Yang, X.; Ma, S.; Yang, A.; Zhang, H.; Yang, S.; Mao, C.; Xu, L.; et al. MiR-34a Targets HDAC1-Regulated H3K9 Acetylation on Lipid Accumulation Induced by Homocysteine in Foam Cells. J. Cell. Biochem. 2017, 118, 4617–4627. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Zeng, L.; Margariti, A.; Xiao, Q.; Li, H.; Zhang, Z.; Pepe, A.E.; Wang, G.; Habi, O.; Defalco, E.; et al. Histone Deacetylase 3 Is Critical in Endothelial Survival and Atherosclerosis Development in Response to Disturbed Flow. Circulation 2010, 121, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.A.; Gijbels, M.J.; Van den Bossche, J.; van der Velden, S.; Sijm, A.; Neele, A.E.; Seijkens, T.; Stoger, J.L.; Meiler, S.; Boshuizen, M.C.; et al. Targeting Macrophage Histone Deacetylase 3 Stabilizes Atherosclerotic Lesions. EMBO Mol. Med. 2014, 6, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mu, H.; Huang, Y.; Li, S.; Wang, Y.; Stetler, R.A.; Bennett, M.V.L.; Dixon, C.E.; Chen, J.; Shi, Y. Microglia-Specific Deletion of Histone Deacetylase 3 Promotes Inflammation Resolution, White Matter Integrity, and Functional Recovery in a Mouse Model of Traumatic Brain Injury. J. Neuroinflamm. 2022, 19, 201. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Hegele, R.A.; Yusuf, S.; Anand, S.S. Multi-Ethnic Genetic Association Study of Carotid Intima-Media Thickness Using a Targeted Cardiovascular SNP Microarray. Stroke 2009, 40, 3173–3179. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and Other Sirtuins in Metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Fry, J.L.; Al Sayah, L.; Weisbrod, R.M.; Van Roy, I.; Weng, X.; Cohen, R.A.; Bachschmid, M.M.; Seta, F. Vascular Smooth Muscle Sirtuin-1 Protects against Diet-Induced Aortic Stiffness. Hypertension 2016, 68, 775–784. [Google Scholar] [CrossRef]
- Li, L.; Gao, P.; Zhang, H.; Chen, H.; Zheng, W.; Lv, X.; Xu, T.; Wei, Y.; Liu, D.; Liang, C. SIRT1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Hypertrophy. Acta Biochim. Biophys. Sin. 2011, 43, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, X.; Zhu, W.; Liu, Z.; Liu, H.; Zhou, Y.; Cao, Y.; Liu, C.; Xie, Y. Resveratrol Enhances Autophagic Flux and Promotes Ox-LDL Degradation in HUVECs via Upregulation of SIRT1. Oxid. Med. Cell. Longev. 2016, 2016, 7589813. [Google Scholar] [CrossRef] [PubMed]
- Gaul, D.S.; Weber, J.; van Tits, L.J.; Sluka, S.; Pasterk, L.; Reiner, M.F.; Calatayud, N.; Lohmann, C.; Klingenberg, R.; Pahla, J.; et al. Loss of Sirt3 Accelerates Arterial Thrombosis by Increasing Formation of Neutrophil Extracellular Traps and Plasma Tissue Factor Activity. Cardiovasc. Res. 2018, 114, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Aneva, I.Y.; Farzaei, M.H.; Sobarzo-Sanchez, E. The Neuroprotective Effects of Astaxanthin: Therapeutic Targets and Clinical Perspective. Molecules 2019, 24, 2640. [Google Scholar] [CrossRef]
- Ambati, R.R.; Moi, P.S.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, Extraction, Stability, Biological Activities and Its Commercial Applications—A Review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Farruggia, C.; Kim, M.B.; Bae, M.; Lee, Y.; Pham, T.X.; Yang, Y.; Han, M.J.; Park, Y.K.; Lee, J.Y. Astaxanthin Exerts Anti-Inflammatory and Antioxidant Effects in Macrophages in NRF2-Dependent and Independent Manners. J. Nutr. Biochem. 2018, 62, 202–209. [Google Scholar] [CrossRef]
- Mendes, K.L.; de Farias Lelis, D.; Santos, S.H.S. Nuclear Sirtuins and Inflammatory Signaling Pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of Poly(ADP-Ribose) Polymerase-1 by P300/CREB-Binding Protein Regulates Coactivation of NF-KappaB-Dependent Transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin Reduces Hepatic Lipid Accumulations in High-Fat-Fed C57BL/6J Mice via Activation of Peroxisome Proliferator-Activated Receptor (PPAR) Alpha and Inhibition of PPAR Gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zarneshan, S.N.; Fakhri, S.; Farzaei, M.H.; Khan, H.; Saso, L. Astaxanthin Targets PI3K/Akt Signaling Pathway toward Potential Therapeutic Applications. Food Chem. Toxicol. 2020, 145, 111714. [Google Scholar] [CrossRef] [PubMed]
- Jourova, L.; Anzenbacherova, E.; Dostal, Z.; Anzenbacher, P.; Briolotti, P.; Rigal, E.; Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Butyrate, a Typical Product of Gut Microbiome, Affects Function of the AhR Gene, Being a Possible Agent of Crosstalk between Gut Microbiome, and Hepatic Drug Metabolism. J. Nutr. Biochem. 2022, 107, 109042. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review Article: The Role of Butyrate on Colonic Function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and Propionate Protect against Diet-Induced Obesity and Regulate Gut Hormones via Free Fatty Acid Receptor 3-Independent Mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Steliou, K.; Boosalis, M.S.; Perrine, S.P.; Sangerman, J.; Faller, D.V. Butyrate Histone Deacetylase Inhibitors. Biores. Open Access 2012, 1, 192–198. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate Inhibits Inflammatory Responses through NFkappaB Inhibition: Implications for Crohn’s Disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate Inhibits NF-ΚB Activation in Lamina Propria Macrophages of Patients with Ulcerative Colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium Butyrate Stimulates Expression of Fibroblast Growth Factor 21 in Liver by Inhibition of Histone Deacetylase 3. Diabetes 2012, 61, 797–806. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate Enhances Mitochondrial Function during Oxidative Stress in Cell Lines from Boys with Autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The Enhancement of Phase 2 Enzyme Activities by Sodium Butyrate in Normal Intestinal Epithelial Cells Is Associated with Nrf2 and P53. Mol. Cell. Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; El Rayess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocińska, K.; Zielińska, D.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front. Pharmacol. 2020, 11, 1–23. [Google Scholar] [CrossRef]
- Shishodia, S. Molecular Mechanisms of Curcumin Action: Gene Expression. Biofactors 2013, 39, 37–55. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The Targets of Curcumin. Curr. Drug Targets 2012, 12, 332–347. [Google Scholar] [CrossRef]
- de Oliveira, G.V.; Alvares, T.S. Effect of Curcumin on Endothelial Function in Humans and Their Proposed Physiological Mechanism: Insights in Formulating Curcumin Products Supplementation. PharmaNutrition 2022, 22, 100313. [Google Scholar] [CrossRef]
- Santos-Parker, J.R.; Strahler, T.R.; Bassett, C.J.; Bispham, N.Z.; Chonchol, M.B.; Seals, D.R. Curcumin Supplementation Improves Vascular Endothelial Function in Healthy Middle-Aged and Older Adults by Increasing Nitric Oxide Bioavailability and Reducing Oxidative Stress. Aging 2017, 9, 187–208. [Google Scholar] [CrossRef]
- Barber-Chamoux, N.; Milenkovic, D.; Verny, M.A.; Habauzit, V.; Pereira, B.; Lambert, C.; Richard, D.; Boby, C.; Mazur, A.; Lusson, J.R.; et al. Substantial Variability Across Individuals in the Vascular and Nutrigenomic Response to an Acute Intake of Curcumin: A Randomized Controlled Trial. Mol. Nutr. Food Res. 2018, 62, 1700418. [Google Scholar] [CrossRef]
- Chiu, J.; Khan, Z.A.; Farhangkhoee, H.; Chakrabarti, S. Curcumin Prevents Diabetes-Associated Abnormalities in the Kidneys by Inhibiting P300 and Nuclear Factor-KappaB. Nutrition 2009, 25, 964–972. [Google Scholar] [CrossRef]
- Tsai, I.J.; Chen, C.W.; Tsai, S.Y.; Wang, P.Y.; Owaga, E.; Hsieh, R.H. Curcumin Supplementation Ameliorated Vascular Dysfunction and Improved Antioxidant Status in Rats Fed a High-Sucrose, High-Fat Diet. Appl. Physiol. Nutr. Metab. 2018, 43, 669–676. [Google Scholar] [CrossRef]
- Choi, Y.; Tanabe, Y.; Akazawa, N.; Zempo-Miyaki, A.; Maeda, S. Curcumin Supplementation Attenuates the Decrease in Endothelial Function Following Eccentric Exercise. J. Exerc. Nutr. Biochem. 2019, 23, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhang, S.; Li, P.; Zheng, X.; Feng, D. Supplementation with Curcumin Inhibits Intestinal Cholesterol Absorption and Prevents Atherosclerosis in High-Fat Diet-Fed Apolipoprotein E Knockout Mice. Nutr. Res. 2018, 56, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.Y.; Zhou, J.; Guo, N.; Ma, W.G.; Huang, X.; Wang, H.; Yuan, Z.Y. Curcumin Retunes Cholesterol Transport Homeostasis and Inflammation Response in M1 Macrophage to Prevent Atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 467, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhu, C.; Ye, S.; Yang, Q. Curcumin Ameliorates Oxidized Low-Density Lipoprotein (Ox-LDL)-Caused Damage in Human Umbilical Vein Endothelial Cells (HUVECs) through the MiR-599/MYD88/NF-KappaB Axis. Toxicol. In Vitro 2022, 85, 105481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ou, C.; Chen, M. Curcumin Attenuates Cadmium-Induced Atherosclerosis by Regulating Trimethylamine-N-Oxide Synthesis and Macrophage Polarization through Remodeling the Gut Microbiota. Ecotoxicol. Environ. Saf. 2022, 244, 114057. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a Novel P300/CREB-Binding Protein-Specific Inhibitor of Acetyltransferase, Represses the Acetylation of Histone/Nonhistone Proteins and Histone Acetyltransferase-Dependent Chromatin Transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef]
- Shishodia, S.; Amin, H.M.; Lai, R.; Aggarwal, B.B. Curcumin (Diferuloylmethane) Inhibits Constitutive NF-KappaB Activation, Induces G1/S Arrest, Suppresses Proliferation, and Induces Apoptosis in Mantle Cell Lymphoma. Biochem. Pharmacol. 2005, 70, 700–713. [Google Scholar] [CrossRef]
- El-Moselhy, M.A.; Taye, A.; Sharkawi, S.S.; El-Sisi, S.F.; Ahmed, A.F. The Antihyperglycemic Effect of Curcumin in High Fat Diet Fed Rats. Role of TNF-Alpha and Free Fatty Acids. Food Chem. Toxicol. 2011, 49, 1129–1140. [Google Scholar] [CrossRef]
- Lin, X.L.; Liu, M.H.; Hu, H.J.; Feng, H.R.; Fan, X.J.; Zou, W.W.; Pan, Y.Q.; Hu, X.M.; Wang, Z. Curcumin Enhanced Cholesterol Efflux by Upregulating ABCA1 Expression through AMPK-SIRT1-LXRalpha Signaling in THP-1 Macrophage-Derived Foam Cells. DNA Cell Biol. 2015, 34, 561–572. [Google Scholar] [CrossRef]
- Kim, J.H. Pharmacological and Medical Applications of Panax Ginseng and Ginsenosides: A Review for Use in Cardiovascular Diseases. J. Ginseng Res. 2018, 42, 264–269. [Google Scholar] [CrossRef]
- Saba, E.; Jeong, D.; Irfan, M.; Lee, Y.Y.; Park, S.J.; Park, C.K.; Rhee, M.H. Anti-Inflammatory Activity of Rg3-Enriched Korean Red Ginseng Extract in Murine Model of Sepsis. Evid. Based Complement. Altern. Med. 2018, 2018, 6874692. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.Y.; Jiang, J.L.; Fang, Y.F.; Yang, F.; Yin, M.D.; Zhang, B.C.; Zhao, R.R.; Shao, J.W. The Effects of Ginsenosides on Platelet Aggregation and Vascular Intima in the Treatment of Cardiovascular Diseases: From Molecular Mechanisms to Clinical Applications. Pharmacol. Res. 2020, 159, 105031. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, S.; Lee, J.Y.; Kim, B. Inhibitory Effects of Ginsenoside Compound K on Lipopolysaccharide-Stimulated Inflammatory Responses in Macrophages by Regulating Sirtuin 1 and Histone Deacetylase 4. Nutrients 2023, 15, 1626. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lim, J.W.; Kim, H. Inhibitory Effect of Korean Red Ginseng Extract on DNA Damage Response and Apoptosis in Helicobacter Pylori–Infected Gastric Epithelial Cells. J. Ginseng Res. 2020, 44, 79–85. [Google Scholar] [CrossRef]
- Qin, M.; Luo, Y.; Lu, S.; Sun, J.; Yang, K.; Sun, G.; Sun, X. Ginsenoside F1 Ameliorates Endothelial Cell Inflammatory Injury and Prevents Atherosclerosis in Mice through A20-Mediated Suppression of NF-KB Signaling. Front. Pharmacol. 2017, 8, 953. [Google Scholar] [CrossRef]
- Lee, A.; Yun, E.; Chang, W.; Kim, J. Ginsenoside Rg3 Protects against IE-DAP-Induced Endothelial-to-Mesenchymal Transition by Regulating the MiR-139-5p-NF-KappaB Axis. J. Ginseng Res. 2020, 44, 300–307. [Google Scholar] [CrossRef]
- Wang, N.; Wan, J.B.; Chan, S.W.; Deng, Y.H.; Yu, N.; Zhang, Q.W.; Wang, Y.T.; Lee, S.M. Comparative Study on Saponin Fractions from Panax Notoginseng Inhibiting Inflammation-Induced Endothelial Adhesion Molecule Expression and Monocyte Adhesion. Chin. Med. 2011, 6, 37. [Google Scholar] [CrossRef]
- Park, J.B.; Kwon, S.K.; Nagar, H.; Jung, S.B.; Jeon, B.H.; Kim, C.S.; Oh, J.H.; Song, H.J.; Kim, C.S. Rg3-Enriched Korean Red Ginseng Improves Vascular Function in Spontaneously Hypertensive Rats. J. Ginseng Res. 2014, 38, 244–250. [Google Scholar] [CrossRef]
- Zhou, P.; Xie, W.; Luo, Y.; Lu, S.; Dai, Z.; Wang, R.; Zhang, X.; Li, G.; Sun, G.; Sun, X. Inhibitory Effects of Ginsenoside Rb1 on Early Atherosclerosis in ApoE-/- Mice via Inhibition of Apoptosis and Enhancing Autophagy. Molecules 2018, 23, 2912. [Google Scholar] [CrossRef]
- Shin, J.H.; Kwon, H.W.; Irfan, M.; Rhee, M.H.; Lee, D.H. Ginsenoside Rk1 Suppresses Platelet Mediated Thrombus Formation by Downregulation of Granule Release and Alpha(IIb)Beta(3) Activation. J. Ginseng Res. 2021, 45, 490–497. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Kim, S.D.; Park, S.C.; Rhee, M.H. Panax Ginseng: Inflammation, Platelet Aggregation, Thrombus Formation, and Atherosclerosis Crosstalk. J. Ginseng Res. 2022, 46, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Qi, Z.; Zheng, B.; Li, Z.; Wang, F.; Liu, J.; Li, P. Inhibitory Effect of Triterpenoids from Panax Ginseng on Coagulation Factor X. Molecules 2017, 22, 649. [Google Scholar] [CrossRef] [PubMed]
- Li, C.T.; Wang, H.B.; Xu, B.J. A Comparative Study on Anticoagulant Activities of Three Chinese Herbal Medicines from the Genus Panax and Anticoagulant Activities of Ginsenosides Rg1 and Rg2. Pharm. Biol. 2013, 51, 1077–1080. [Google Scholar] [CrossRef]
- Kim, M.H.; Lee, J.; Jung, S.; Kim, J.W.; Shin, J.H.; Lee, H.J. The Involvement of Ginseng Berry Extract in Blood Flow via Regulation of Blood Coagulation in Rats Fed a High-Fat Diet. J. Ginseng Res. 2017, 41, 120–126. [Google Scholar] [CrossRef]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC Inhibitors: Targets for Tumor Therapy, Immune Modulation and Lung Diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Kim, H.S.; Kim, D.H.; Bae, S.C.; Hyun, J.W. Compound K, a Metabolite of Ginseng Saponin, Inhibits Colorectal Cancer Cell Growth and Induces Apoptosis through Inhibition of Histone Deacetylase Activity. Int. J. Oncol. 2013, 43, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Li, J.; Xia, J.; Jiang, R.; Zuo, G.W.; Li, X.P.; Chen, Y.; Xiong, W.; Chen, D.L. Ginsenoside 20(s)-Rh2 as Potent Natural Histone Deacetylase Inhibitors Suppressing the Growth of Human Leukemia Cells. Chem. Biol. Interact. 2015, 242, 227–234. [Google Scholar] [CrossRef]
- Shan, X.; Fu, Y.S.; Aziz, F.; Wang, X.Q.; Yan, Q.; Liu, J.W. Ginsenoside Rg3 Inhibits Melanoma Cell Proliferation through Down-Regulation of Histone Deacetylase 3 (HDAC3) and Increase of P53 Acetylation. PLoS ONE 2014, 9, e115401. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Yu, L.; Redpath, P.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is a Major NAD+ Precursor Vitamin in Cow Milk. J. Nutr. 2016, 146, 957–963. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Zhang, T.; Kraus, W.L. SIRT1-Dependent Regulation of Chromatin and Transcription: Linking NAD+ Metabolism and Signaling to the Control of Cellular Functions. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide Riboside Attenuates Alcohol Induced Liver Injuries via Activation of SirT1/PGC-1alpha/Mitochondrial Biosynthesis Pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-Mediated Biological Control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient Control of Glucose Homeostasis through a Complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Nishimuro, H.; Ohnishi, H.; Sato, M.; Ohnishi-Kameyama, M.; Matsunaga, I.; Naito, S.; Ippoushi, K.; Oike, H.; Nagata, T.; Akasaka, H.; et al. Estimated Daily Intake and Seasonal Food Sources of Quercetin in Japan. Nutrients 2015, 7, 2345–2358. [Google Scholar] [CrossRef]
- Kobori, M.; Takahashi, Y.; Sakurai, M.; Akimoto, Y.; Tsushida, T.; Oike, H.; Ippoushi, K. Quercetin Suppresses Immune Cell Accumulation and Improves Mitochondrial Gene Expression in Adipose Tissue of Diet-Induced Obese Mice. Mol. Nutr. Food Res. 2016, 60, 300–312. [Google Scholar] [CrossRef]
- Lu, X.L.; Zhao, C.H.; Yao, X.L.; Zhang, H. Quercetin Attenuates High Fructose Feeding-Induced Atherosclerosis by Suppressing Inflammation and Apoptosis via ROS-Regulated PI3K/AKT Signaling Pathway. Biomed. Pharmacother. 2017, 85, 658–671. [Google Scholar] [CrossRef]
- Xiao, L.; Liu, L.; Guo, X.; Zhang, S.; Wang, J.; Zhou, F.; Liu, L.; Tang, Y.; Yao, P. Quercetin Attenuates High Fat Diet-Induced Atherosclerosis in Apolipoprotein E Knockout Mice: A Critical Role of NADPH Oxidase. Food Chem. Toxicol. 2017, 105, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Weng, X.; Bao, X.; Bai, X.; Lv, Y.; Zhang, S.; Chen, Y.; Zhao, C.; Zeng, M.; Huang, J.; et al. A Novel Anti-Atherosclerotic Mechanism of Quercetin: Competitive Binding to KEAP1 via Arg483 to Inhibit Macrophage Pyroptosis. Redox Biol. 2022, 57, 102511. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.-L.; Sheng, Y.-C.; Zheng, Z.-Y.; Shi, L.; Wang, Z.-T. The Involvement of P62–Keap1–Nrf2 Antioxidative Signaling Pathway and JNK in the Protection of Natural Flavonoid Quercetin against Hepatotoxicity. Free Radic. Biol. Med. 2015, 85, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Xiang, L.; Xiao, L. Quercetin Alleviates Atherosclerosis by Suppressing Oxidized LDL-Induced Senescence in Plaque Macrophage via Inhibiting the P38MAPK/P16 Pathway. J. Nutr. Biochem. 2023, 116, 109314. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, F.; Qian, X.; Luo, S.; Wang, Z.; Gao, X.; Kong, X.; Zhang, J.; Chen, S. Quercetin Protects Endothelial Function from Inflammation Induced by Localized Disturbed Flow by Inhibiting NRP2 -VEGFC Complex. Int. Immunopharmacol. 2023, 116, 109842. [Google Scholar] [CrossRef]
- Hung, C.H.; Chan, S.H.; Chu, P.M.; Tsai, K.L. Quercetin Is a Potent Anti-Atherosclerotic Compound by Activation of SIRT1 Signaling under OxLDL Stimulation. Mol. Nutr. Food Res. 2015, 59, 1905–1917. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.-J.; Qian, H.-Y.; Zhang, Q.; Xu, H.; Li, J.-J. Resveratrol in Cardiovascular Disease: What Is Known from Current Research? Heart Fail. Rev. 2012, 17, 437–448. [Google Scholar] [CrossRef]
- Guo, S.; Zhou, Y.; Xie, X. Resveratrol Inhibiting TGF/ERK Signaling Pathway Can Improve Atherosclerosis: Backgrounds, Mechanisms and Effects. Biomed. Pharmacother. 2022, 155, 113775. [Google Scholar] [CrossRef]
- Zhang, L.X.; Li, C.X.; Kakar, M.U.; Khan, M.S.; Wu, P.F.; Amir, R.M.; Dai, D.F.; Naveed, M.; Li, Q.Y.; Saeed, M.; et al. Resveratrol (RV): A Pharmacological Review and Call for Further Research. Biomed. Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef]
- Lan, D.; Lu, M.; Sharma, S.; Mellon, P.L.; Olefsky, J.M.; Webster, N.J.G. Trans-Resveratrol Inhibits Phosphorylation of Smad2/3 and Represses FSHβ Gene Expression by a SirT1-Independent Pathway in LβT2 Gonadotrope Cells. Reprod. Toxicol. 2011, 32, 85–92. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Zeng, M.; Zhang, Y.; Bu, P. Activation of SIRT3 by Resveratrol Ameliorates Cardiac Fibrosis and Improves Cardiac Function via the TGF-Beta/Smad3 Pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H424–H434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, Y.; Ong’achwa, M.J.; Ge, L.; Qian, Y.; Chen, L.; Hu, X.; Li, F.; Wei, H.; Zhang, C.; et al. Resveratrol Inhibits the TGF-Beta1-Induced Proliferation of Cardiac Fibroblasts and Collagen Secretion by Downregulating MiR-17 in Rat. Biomed. Res. Int. 2018, 2018, 8730593. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Luo, J.Y.; Lau, C.W.; Chen, Z.Y.; Tian, X.Y.; Huang, Y. Pharmacological Basis and New Insights of Resveratrol Action in the Cardiovascular System. Br. J. Pharmacol. 2020, 177, 1258–1277. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.; Thandapilly, S.J.; Wigle, J.; Zieroth, S.; Netticadan, T. A Comprehensive Analysis of the Efficacy of Resveratrol in Atherosclerotic Cardiovascular Disease, Myocardial Infarction and Heart Failure. Molecules 2021, 26, 6600. [Google Scholar] [CrossRef]
- Dong, W.; Wang, X.; Bi, S.; Pan, Z.; Liu, S.; Yu, H.; Lu, H.; Lin, X.; Wang, X.; Ma, T.; et al. Inhibitory Effects of Resveratrol on Foam Cell Formation Are Mediated through Monocyte Chemotactic Protein-1 and Lipid Metabolism-Related Proteins. Int. J. Mol. Med. 2014, 33, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Liu, J.L.; Du, A.L. Cardioprotective Effect of Resveratrol on Atherogenic Diet-Fed Rats. Int. J. Clin. Exp. Pathol. 2014, 7, 7899–7906. [Google Scholar]
- Deng, Z.Y.; Hu, M.M.; Xin, Y.F.; Gang, C. Resveratrol Alleviates Vascular Inflammatory Injury by Inhibiting Inflammasome Activation in Rats with Hypercholesterolemia and Vitamin D2 Treatment. Inflamm. Res. 2015, 64, 321–332. [Google Scholar] [CrossRef]
- Thompson, A.M.; Martin, K.A.; Rzucidlo, E.M. Resveratrol Induces Vascular Smooth Muscle Cell Differentiation through Stimulation of SirT1 and AMPK. PLoS ONE 2014, 9, e85495. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Tang, W.; Qiu, Q.; Peng, J. Resveratrol Prevents TNF-α-Induced VCAM-1 and ICAM-1 Upregulation in Endothelial Progenitor Cells via Reduction of NF-ΚB Activation. J. Int. Med. Res. 2020, 48, 0300060520945131. [Google Scholar] [CrossRef]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.J.; et al. Resveratrol Attenuates Vascular Endothelial Inflammation by Inducing Autophagy through the CAMP Signaling Pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a Promising Molecule for Fighting Cancer. Mutat. Res. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary Sulforaphane, a Histone Deacetylase Inhibitor for Cancer Prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Kee, H.J.; Jin, L.; Ryu, Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Inhibition of Class IIa Histone Deacetylase Activity by Gallic Acid, Sulforaphane, TMP269, and Panobinostat. Biomed. Pharmacother. 2018, 101, 145–154. [Google Scholar] [CrossRef]
- Tortorella, S.M.; Royce, S.G.; Licciardi, P.V.; Karagiannis, T.C. Dietary Sulforaphane in Cancer Chemoprevention: The Role of Epigenetic Regulation and HDAC Inhibition. Antioxid. Redox Signal. 2015, 22, 1382–1424. [Google Scholar] [CrossRef]
- Sharma, M.; Tollefsbol, T.O. Combinatorial Epigenetic Mechanisms of Sulforaphane, Genistein and Sodium Butyrate in Breast Cancer Inhibition. Exp. Cell Res. 2022, 416, 113160. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone Deacetylase Turnover and Recovery in Sulforaphane-Treated Colon Cancer Cells: Competing Actions of 14-3-3 and Pin1 in HDAC3/SMRT Corepressor Complex Dissociation/Reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A Novel Mechanism of Chemoprotection by Sulforaphane: Inhibition of Histone Deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Borsheim, E.; Jun, S.R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane Prevents Age-Associated Cardiac and Muscular Dysfunction through Nrf2 Signaling. Aging Cell 2020, 19, e13261. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Z.; Yang, Q.; Li, H.; Guan, H.; Shi, B.; Hou, P.; Ji, M. Sulforaphane Inhibits Thyroid Cancer Cell Growth and Invasiveness through the Reactive Oxygen Species-Dependent Pathway. Oncotarget 2015, 6, 25917–25931. [Google Scholar] [CrossRef]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane Enhances Nrf2 Expression in Prostate Cancer TRAMP C1 Cells through Epigenetic Regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H. Regulation of Acetylation States by Nutrients in the Inhibition of Vascular Inflammation and Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 9338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119338
Kang H. Regulation of Acetylation States by Nutrients in the Inhibition of Vascular Inflammation and Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(11):9338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119338
Chicago/Turabian StyleKang, Hyunju. 2023. "Regulation of Acetylation States by Nutrients in the Inhibition of Vascular Inflammation and Atherosclerosis" International Journal of Molecular Sciences 24, no. 11: 9338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119338