4. Materials and Methods
4.1. General
NMR spectra were recorded using the Varian spectrometers (Darmstadt, Germany) DD2 and VNMRS (400 and 500 MHz, respectively). MS spectra were taken on an Advion expressionL CMS mass spectrometer (Ithaca, NY, USA; positive ion polarity mode, solvent: methanol, solvent flow: 0.2 mL/min, spray voltage: 5.17 kV, source voltage: 77 V, APCI corona discharge: 4.2 μA, capillary temperature: 250 °C, capillary voltage: 180 V, sheath gas: N2). Thin-layer chromatography was performed on precoated silica gel plates supplied by Macherey-Nagel (Düren, Germany). IR spectra were recorded on a Spectrum 1000 FT-IR-spectrometer from Perkin Elmer (Rodgau, Germany). The UV/Vis-spectra were recorded on a Lambda 14 spectrometer from Perkin Elmer (Rodgau, Germany); optical rotations were measured at 20 °C using a JASCO-P2000 instrument (JASCO Germany GmbH, Pfungstadt, Germany). The melting points (m.p.) were determined using the Leica hot-stage microscope Galen III (Leica Biosystems, Nussloch, Germany) and are uncorrected. The solvents were dried according to the usual procedures. Microanalyses were performed with an Elementar Vario EL (CHNS) instrument (Elementar Analysensysteme GmbH, Elementar-Straße 1, D-63505, Langenselbold, Germany).
All dry solvents were distilled over respective drying agents except for DMF which was distilled and stored under argon and a molecular sieve. Reactions using air- or moisture-sensitive reagents were carried out under an argon atmosphere in dried glassware. Triethylamine was stored over potassium hydroxide. Biological assays were performed as previously reported. The parent triterpenoic acids were obtained from local vendors.
4.2. General Procedure for Acetylation (GP 1)
To a solution of the parent triterpenoic acid (1 equiv.) in dry DCM, acetic anhydride (3 equiv.), dry triethylamine (3 equiv.), and DMAP (catal. amounts) were added, and the mixture was stirred at 20 °C for one day. The usual aqueous work-up followed by re-crystallization from ethanol furnished the corresponding acetates 9–13. Their respective m.p., values, 1H, and 13C NMR spectra, as well as ESI MS data, correspond to the literature values.
4.3. General Procedure for the Synthesis of Amides 14–18 (GP 2)
To a solution of acetates 9–13 (1 equiv.) in dry DCM (100 mL), oxalyl chloride (5 equiv.) and DMF (2 drops) were added and the mixture was stirred at 20 °C for 2 h. The volatiles were removed under diminished pressure and the residue was dissolved in dry DCM (100 mL). This solution was slowly added to a solution of the corresponding amine (3 equiv.) in dry acetonitrile (100 mL) in the presence of DMAP (catal. amounts). The mixture was stirred at 20 °C for 1 day, the volatiles were removed under diminished pressure, and the residue was subjected to column chromatography (silica gel) to afford products 14–18.
4.4. General Procedure for the Synthesis of the Rhodamine Conjugates 19–28 (GP 3)
To a solution of the rhodamine (rhodamine B or rhodamine 101, 1 equiv.) in dry DCM (100 mL), oxalyl chloride (7 equiv.) and dry DMF (2 drops) were added, and the mixture was stirred at 20 °C for 1 h. The volatiles were removed under diminished pressure and the residue was dissolved in dry DCM (100 mL). A solution of the corresponding amine (1 equiv.) in dry DCM (100 mL) was added, followed by the addition of catal. amounts of triethylamine and DMAP. The mixture was stirred at 20 °C for 1 h (TLC showed completion of the reaction), the solvents were removed in vacuo, and the residue was subjected to column chromatography (silica gel, CHCl3/MeOH) to afford products 19–28.
4.5. N,N’-Ditosyl-1,3-propanediamine (5)
Tosyl chloride (40.0 g, 210 mmol) was molten in a beaker at 80 °C and 1,3-propanediamine (3, 8.9 mL, 106 mmol) was added dropwise; to complete the reaction, the mixture was stirred for an additional 30 min at 80 °C. After cooling to 20 °C, aq. HCl (2
m) was added, and the precipitate was washed with water followed by a recrystallization from ethanol to furnish
5 (33.7 g, 83%) as a colorless solid; m.p. 138 °C (lit: [
49] 137–140 °C); R
f = 0.75 (silica gel, hexanes/ethyl acetate, 4:6); UV-Vis (CHCl
3): λ
max (log ε) = 228 nm (4.16); IR (ATR):
ν = 3271
w, 1595
w, 1431
w, 1305
s, 1214
w, 1154
s, 1088
m, 1024
w, 980
m, 858
m, 815
s, 698
s, 550
s, 568
s, 489
m cm
−1;
1H NMR (400 MHz, CDCl
3): δ = 7.73 (
m, 4H, 4-H, 8-H), 7.32–7.25 (
m, 4H, 5-H, 7-H), 3.02 (
t,
J = 5.8 Hz, 4H, 2-H), 2.42 (
s, 6H, 9-H), 1.67 (
p,
J = 6.2 Hz, 2H, 1-H) ppm;
13C NMR (101 MHz, CDCl
3): δ = 143.6 (C-6), 136.8 (C-3), 129.8 (C-5, C-7), 127.0 (C-4, C-8), 39.8 (C-2), 29.9 (C-1), 21.5 (C-9) ppm; MS (ESI, MeOH/CHCl
3, 4:1):
m/
z = 405.0 (100%, [M+Na]
+).
4.6. 1,3-Propanediol Ditosylate (6)
A mixture of 1,3-propanediol (
4, 16.0 g, 210 mmol) and tosyl chloride (88.0 g, 461 mmol) in dry pyridine (70 mL) was stirred at 0 °C for 1 h. The product was precipitated by adding aq. HCl (2
m), filtered off and dried. Compound
6 (69.9 g, 87%) was obtained as a colorless solid; m.p. 92 °C (lit.: [
51] 92–93 °C); R
f = 0.49 (hexanes/ethyl acetate, 6:4); UV-Vis (CHCl
3): λ
max (log ε) = 225 nm (4.11); IR (ATR):
ν = 2978
w, 1599
m, 1496
w, 1470
w, 1421
w, 1352
s, 1293
m, 1254
w, 1190
m, 1172
s, 1095
m, 1029
m, 1021
m, 941
s, 892
w, 852
s, 810
s, 739
s, 660
s, 580
s, 568
s, 549
s, 488
m cm
−1;
1H NMR (400 MHz, CDCl
3): δ = 7.75–7.23 (
m, 8H, 4-H, 5-H, 7-H, 8-H), 4.06 (
t,
J = 6.0 Hz, 4H, 2-H), 2.46 (s, 6H, 9-H), 1.99 (
p,
J = 6.0 Hz, 2H, 1-H) ppm;
13C NMR (101 MHz, CDCl
3): δ = 145.1 (C-6), 132.6 (C-3), 130.0 (C-5, C-7), 127.9 (C-4, C-8), 65.9 (C-2), 28.7 (C-1), 21.6 (C-9) ppm; MS (ESI, MeOH/CHCl
3, 4:1):
m/
z = 407.3 (100%, [M+Na]
+).
4.7. 1,5-Bis (p-Toluenesulfonyl)-1,5-diazacyclooctane (7)
To a solution of sodium methanolate (8.0 g, 148 mmol) in dry MeOH (100 mL)
5 (5.0 g, 13 mmol) was added, and the mixture was heated under reflux for 4 h. The solvent was removed, the residue was dissolved in dry DMF (100 mL) and 6 (5.0 g, 13 mmol) was added. The mixture was stirred at 80 °C for 12 h. The product was precipitated by adding aq. HCl (2
m), filtered off, and
7 (4.7 g, 84%) was obtained as a colorless solid; m.p. 214–216 °C (lit. [
33]: 214–215 °C); R
f = 0.33 (hexane/ethyl acetate, 7:3); UV-Vis (CHCl
3): λ
max (log ε) = 232 nm (4.32); IR (ATR):
ν = 2953
w, 1597
w, 1456
m, 1378
m, 1321
s, 1182
m, 1150
s, 1088
s, 1017
m, 1059
s, 987
s, 927
m, 837
m, 812
s, 723
s, 644
s, 627
m, 543
s, 487
m, 462
m, 408
m cm
−1;
1H NMR (400 MHz, CDCl
3): δ = 7.68 (
d,
J = 8.3 Hz, 4H, 5-H, 9-H), 7.33–7.30 (
m, 4H, 6-H, 8-H), 3.31–3.24 (
m, 8H, 1-H, 3-H), 2.43 (
s, 6H, 10-H), 2.04 (
p,
J = 5.9 Hz, 4H, 2-H) ppm;
13C NMR (101 MHz, CDCl
3): δ = 143.4 (C-7), 135.6 (C-4), 129.8 (C-6, C-8), 127.1 (C-5, C-9), 47.0 (C-1, C-3), 30.2 (C-2), 21.5 (C-10) ppm; MS (ESI, MeOH/CHCl
3, 4:1):
m/
z = 445.2 (100%, [M+Na]
+).
4.8. 1,5-Diazacyclooctane Dihydrobromide (8)
4.8.1. Procedure A
A solution of 7 (2.5 g, 6 mmol) and thioanisole (2.4 mL, 18 mmol) in HBr (33% in glacial acetic acid, 150 mL) was stirred at 80 °C for 3 h. The volatiles were removed under diminished pressure, DCM (30 mL) was added, and the solution was washed with water (3 × 100 mL), followed by decolorization (activated charcoal). The solution was filtered, the solvent removed, and 8 (1.5 g, 5.5 mmol, 92%) was obtained as a colorless solid.
4.8.2. Procedure B
A solution of hydrazine (75 mL, 1.5 mol) in EtOH (200 mL) was heated under reflux, and 1,3-dibromopropane (75 mL, 0.75 mol) was added slowly within 4 h. Stirring was continued for another hour, the solids were filtered off, washed with ethanol (3 × 50 mL), and discarded. The pH of the filtrate [combined with the EtOH washings and additional water (150 mL)] was adjusted to pH =3 by adding aqu. HBr (48% in water). Benzaldehyde (60 mL, 0.6 mol) was added, and the precipitate formed upon addition was filtered off, washed with water (3 × 50 mL), and discarded. The combined filtrates were extracted with ether (1000 mL), and the aq. The layer was concentrated under diminished pressure resulting in the formation of a red solid. Ethanol (250 mL) was added, and shaking of this suspension was continued for another 5 min. The yellowish solid was filtered off, washed with ethanol (250 mL) and ether (5 × 100 mL), and
8 (15.6 g, 7.5%) was obtained as a colorless solid; m.p. = 220–225 °C (lit.: [
51,
52] >250 °C); R
f = 0.8 (CHCl
3:MeOH, 95:5); IR (ATR):
ν = 2971
s, 2728
s, 2418
m, 1577
s, 1461
s, 1331
m, 1095
s, 1027
m, 890
m, 696
m, 547
m, 491
m, cm
−1;
1H NMR (400 MHz, D
2O): δ = 3.36–3.31 (
m, 8H, 1-H, 3-H, 4-H, 6-H), 2.22–2.16 (
m, 4H, 2-H, 5-H) ppm;
13C NMR (101 MHz, D
2O): δ = 43.8 (C-1, C-3, C-4, C-6), 20.8 (C-2, C-5) ppm; MS (ESI, MeOH/CHCl
3, 4:1):
m/
z = 115.0 (100%, [M+H-2 HBr]
+).
4.9. (3ß)28-(1,5-Diazocan-1-yl)-28-oxoolean-12-en-3-yl Acetate (14)
Following GP 2 from 3-O-acetyl-oleanolic acid (9, 500 mg, 1.0 mmol), followed by chromatography (silica gel, CHCl3/MeOH (2% → 10%), compound 14 (425 mg, 71%) was obtained as a colorless solid; m.p. = 207–210 °C (decomp.); Rf = 0.52 (CHCl3/MeOH, 95:5); = + 3.8° (c 0.088, CHCl3); IR (ATR): ν = 2954m, 1732s, 1626m, 1464m, 1368s, 1245s, 1026s, 750s, 662w cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.25 (m, 1H, 12-H), 4.50 (m, 1H, 3-H), 3.67–3.11 (m, 8H, 33-H, 35-H, 36-H, 38-H), 3.03 (d, J = 13.8 Hz 1H, 18-H), 2.16–2.12 (m, 1H, 16-H), 2.04 (s, 3H, 32-H), 1.87–1.17 (m, 23H, 11-H, 34-H, 37-H, 19-Ha, 2-H, 1-Ha, 9-H, 6-Ha, 15-H, 7-H, 21-H, 6 Hb, 22-H, 19-Hb), 1.13 (s, 3H, 27-H), 1.01–0.98 (m, 1H, 1-Hb), 0.97-0.93 (s, 3H, 25-H), 0.92 (s, 3H, 30-H), 0.89 (s, 3H, 29-H), 0.85 (s, 3H, 23-H), 0.84 (s, 3H, 24-H), 0.82-0.81 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 177.0 (C-28), 171.2 (C-31), 144.7 (C-13), 121.7 (C-12), 81.1 (C-3), 55.5 (C-5), 47.9 (C-33, C-35, C-36, C-38), 47.8 (C-9),47.4 (C-17), 46.6 (C-19), 43.9 (C-18), 42.6 (C-14), 39.2 (C-8), 38.2 (C-1), 37.8 (C-4), 37.1 (C-10), 34.2 (C-21), 33.1 (C-7), 33.0 (C-29), 30.5 (C-34, C-37), 30.4 (C-20),29.8 (C-22), 28.2 (C-23), 27.6 (C-15), 26.0 (C-27), 24.1 (C-30), 23.7 (C-2), 23.5 (C-11), 22.8 (C-16), 21.4 (C-32), 18.3 (C-6), 17.3 (C-26), 16.8 (C-24), 15.6 (C-25) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 596.3 (100%, [M+H]+); analysis calcd. for C38H62N2O3 (594.93): C 76.72, H 10.50, N 4.71; found: C 76.47, H 10.74, N 4.50.
4.10. (3ß) 28-(1,5-Diazocan-1-yl)-28-oxours-12-en-3-yl Acetate (15)
Following GP 2 from 3-O-acetyl-ursolic acid (10, 500 mg, 1.0 mmol), followed by chromatography (silica gel, CHCl3/MeOH (2% → 10%), compound 15 (413 mg, 69%) was obtained as an off-white solid; m.p. = 232–235 °C (decomp.); Rf = 0.37 (CHCl3/MeOH, 95:5); = + 0.45° (c 0.088, CHCl3); IR (ATR): ν = 2942m, 1731m, 1627m, 1456m, 1370s, 1245s, 1026s, 750s, 662m cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.18–5.13 (m, 1H, 12-H), 4.46–4.39 (m, 1H, 3-H), 3.74–3.01 (m, 8H, 33-H, 35-H, 36-H, 38-H), 2.39 (d, J = 11.3 Hz, 1H, 18-H), 1.99 (s, 3H, 32-H), 1.88–1.82 (m, 2H, 11-H), 1.74–1.67 (m, 1H, 20-H), 1.73–1.05 (m, 23H, 2-H, 6-H, 15-H, 16-H, 21-H, 7-H, 9-H, 22-H, 1Ha, 19-H, 34-H, 37-H), 1.02 (s, 3H, 27-H), 0.99–0.95 (m, 1H, 1Hb), 0.92 (s, 3H, 23-H), 0.88 (s, 3H, 30-H), 0.86 (s, 3H, 25-H), 0.81 (s, 3H, 29-H), 0.80 (s, 3H, 24-H), 0.77-0.74 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 174.9 (C-28), 171.0 (C-31), 125.3 (C-12), 80.9 (C-3), 55.2 (C-5), 55.0 (C-18), 48.7 (C-33, C-35, C-36, C-38), 48. 6 (C-17), 47.7 (C-9), 43.4 (C- 8), 43.5 (C-14), 39.4 (C-19), 38.7 (C-20), 38.6 (C-1), 37.6 (C-4), 37.0 (C-10), 33.9 (C-22), 32.9 (C-7), 30.6 (C-21), 28.1 (C-23), 27.3 (C-34, C-37), 27.0 (C-15), 26.4 (C-16), 23,4 (C-27), 23.5 (C-2), 23.3 (C-11), 21.2 (C-32), 21.0 (C-30), 18.3 (C-6), 16.7 (C-29), 16.39 (C-26), 15.62 (C-24), 15.40 (C-25) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 596.2 (100%, [M+H]+); analysis calcd. for C38H62N2O3 (594.93): C 76.72, H 10.50, N 4.71; found: C 76.58, H 10.76, N 4.49.
4.11. (3ß) 28-(1,5-Diazocan-1-yl)-28-oxolup-20(29)-en-3-yl Acetate (16)
Following GP 2 from 3-O-acetyl-betulinic acid (11, 500 mg, 1.0 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH (10% → 50%), compound 16 (430 mg, 72%) was obtained as a colorless solid; m.p. 223–234 °C (decomp.); Rf = 0.43 (CHCl3/MeOH, 9:1); = −8.0° (c 0.064, CHCl3); IR (ATR): ν = 3408w, 2942m, 1731m, 1632s, 1455m, 1373s, 1246s, 1195m, 1026m, 979m, 882m, 730s cm−1; 1H NMR (400 MHz, CDCl3): δ = 4.68 (m, 1H, 29-Ha), 4.56–4.53 (m, 1H, 29-Hb), 4.42 (dd, J = 10.1, 6.2 Hz, 1H, 3-H), 4.02–3.19 (m, 8H, 33-H, 35-H, 36-H, 38-H), 2.85 (m, 2H, 13-H, 19-H), 2.13–2.08 (m, 1H, 16-Ha), 2.00 (s, 3H, 32-H), 1.96–1.93 (m, 1H, 22-Ha), 1.78–1.74 (m, 1H, 21-Ha1.65–1.63 (m, 5H, 1-Ha, 12-Ha, 30-H), 1.60–1.05 (m, 2-H, 16-Hb, 18-H, 6-Ha, 7-H, 21-H, 11-H, 22-Hb,34-H, 37-H, 9-H, 15-H), 0.96–0.94 (m, 1H, 1-Hb), 0.92 (s, 3H, 27-H), 0.90–0.89 (m, 1H, 12-Hb), 0.87 (s, 3H, 25-H), 0.80 (s, 3H,m 24-H), 0.79 (s, 3H, 23-H), 0.76–0.74 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 175.4 (C-28), 171.0 (C-31), 150.8 (C-20), 109.4 (C-29), 80.9 (C-3), 55.5 (C-5), 55.2 (C-33, C-35, C-36, C-38), 55.0 (C-17), 52.9 (C-18), 50.7 (C-9), 45.6 (C-19), 42.0 (C-14), 40.7 (C-8), 38.8 (C-4), 38.4 (C-1), 37.8 (C-10), 37.1 (C-7), 36.9 (C-13), 36.1 (C-22), 34.3 (C-34, C-37), 32.3 (C-16), 31.4 (C-21), 30.1 (C-15), 25.5 (C-23), 23.7 (C-12), 23.7 (C-2), 21.3 (C-32), 21.1 (C-11), 19.7 (C-30), 18.1 (C-6), 16.4 (C-24), 16.2 (C-25), 16.0 (C-26), 14.6 (C-27) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 596.1 (100%, [M+H]+); analysis calcd. for C38H62N2O3 (594.93): C 76.72, H 10.50, N 4.71; found: C 76.46, H 10.77, N 4.53.
4.12. (3ß)28-(1,5-Diazocan-1-yl)-30-nor-20,28-dioxolup-3-yl-acetate (17)
Following GP 2 from 3-O-acetyl-platanic acid (12, 500 mg, 1.0 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH (10% → 50%), compound 17 (425 mg, 70%) was obtained as a colorless solid; m.p. = 210–214 °C (decomp.); Rf = 0.44 (CHCl3/MeOH, 9:1); = −26.6° (c 0.028, CHCl3); IR (ATR): ν = 3396w, 2942m, 2866m, 1731m, 1626s, 1466m, 1411m, 1369m, 1197s, 1245m, 1120m, 1025m, 978m cm−1; 1H NMR (400 MHz, CDCl3): δ = 4.39 (dd, J = 10.6, 5.5 Hz, 1H, 3-H), 3.78–3.06 (m, 9H, 19-H, 32-H, 34-H, 35-H, 37-H), 2.66–2.56 (m, 1H, 13-H), 2.10 (s, 3H, 29-H), 2.08–1.98 (m, 2H, 16-Ha, 18-H), 1.97 (s, 3H, 31-H), 1.94–1.90 (m, 1H, 22-Ha), 1.82–1.76 (m, 1H, 21-Ha), 1.70–1.05 (m, 19H, 1-Ha, 16-Hb, 2-H, 22-Hb, 21-Hb, 6-Ha, 11-Ha, 7-H, 6Hb, 9-H, 15-H, 11-Hb, 33-H, 36-H), 0.98–0.95 (m, 2H, 12-H), 0.92 (s, 3H, 27-H), 0.91–0.85 (m, 1H, 1-Hb), 0.83 (s, 3H, 24-H), 0.79–0.77 (m, 6H, 23-H, 25-H), 0.76 (s, 3H, 26-H), 0.72–0.71 (m, 1H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 212.5 (C-20), 175.3 (C-28), 170.9 (C-30), 80.8 (C-3), 55.4 (C-5), 55.1 (C-32, C-34, C-35, C-37), 52.7 (C-18), 50.6 (C-9), 49.9 (C-19), 46.1 (C-17), 41.8 (C-8), 40.6 (C-14), 38.3 (C-1), 37.7 (C-4), 37.1 (C-10), 35.9 (C-13), 35.8 (C-22), 34.1 (C-7), 31.8 (C-16), 30.3 (C-29), 30.0 (C-15), 28.8 (C-21), 27.9 (C-23), 27.3 (C-12), 23.6 (C-2), 21.3 (C-31), 21.1 (C-11), 18.1 (C-6), 16.4 (C-26), 16.2 (C-25), 15.9 (C-24), 14.6 (C-27) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 597.3 (100%, [M+H+); analysis calcd. for C37H60N2O4 (596.90): C 74.45, H 10.13, N 4.69; found: C 74.21, H 10.32, N 4.43.
4.13. (2α,3ß,4α)28-(1,5-Diazocan-1-yl)-28-oxours-12-ene-2,3,23-triyl Triacetate (18)
Following GP 2 from 2,3,24-tri-O-acetyl-asiatic acid (13, 400 mg, 0.8 mmol), followed by chromatography [silica gel, CHCl3/MeOH (2% → 50%)], compound 18 (425 mg, 74%) was obtained as colorless solid; m.p. = 187–190 °C (decomp.); Rf = 0.38 (CHCl3/MeOH, 9:1); = −30.2° (c 0.015, CHCl3); IR (ATR): ν = 2925w, 1741s, 1623w, 1368m, 1231s, 1042m, 748w cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.22–5.17 (m, 1H, 12-H), 5.14–5.08 (m, 1H, 2-H), 5.04–5.01 (m, 1H, 3-H), 3.80 (m, 1H, 23-Ha), 3.51 (m, 1H, 23-Hb), 3.32–2.67 (m, 8H, 37-H, 39-H, 40-H, 42-H), 2.40–2.34 (m, 1H, 18-H), 2.03 (s, 3H, 36-H), 2.02–2.00 (m, 1H, 1-Ha), 1.97 (s, 3H, 34-H), 1.92 (s, 3H, 32-H), 1.88–1.69 (m, 5H, 11-H, 16-H 22-Ha), 1.60–1.45 (m, 4H, 22-Hb, 9-H, 21-Ha, 7-Ha), 1.53–1.45 (m, 2H, 16-Ha, 16-Hb), 1.33–1.14 (m, 10H, 19-H, 5-H, 21-Hb, 7-Hb, 15-H, 38-H, 41-H), 1.11–1.09 (m, 1H, 1-Hb), 1.05 (s, 3H, 27-H), 1.02 (s, 3H, 25-H), 0.99-0.96 (m, 1H, 20-H), 0.91 (s, 3H, 30-H), 0.84 (s, 3H, 24-H), 0.82 (s, 3H, 29-H), 0.70 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 176.8 (C-28), 170.8 (C-35), 170.3 (C-33), 170.3 (C-31), 138.7 (C-13), 124.9 (C-12), 74.8 (C-3), 69.8 (C-2), 65.2 (C-23), 55.6 (C-18), 47.6 (C-9), 47.5 (C-5), 46.1 (C-37, C-39, C-40, C-42), 43.7 (C-1), 42.3 (C-14), 41.9 (C-4), 39.5 (C-8), 39.3 (C-19), 38.5 (C-20), 37.8 (C-10), 34.8 (C-22), 34.7 (C-7), 31.9 (C-21), 29.6 (C-15), 23.3 (C-11), 23.2 (C-16), 22.6 (C-38, C-41), 21.2 (C-30), 21.0 (C-36), 20.8 (C-32), 20.7 (C-34), 17.8 (C-6), 17.4 (C-27), 17.3 (C-29), 17.1 (C-25), 13.9 (C-26), 8.7 (C-24) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 711.8 (100%, [M+H+); analysis calcd. for C42H66N2O7 (711.00): C 70.95, H 9.36, N 3.94; found: C 70.69, H 9.51, N 3.75.
4.14. (3ß)-3-Acetyloxy-28-(5-{2-[3,6-bis(diethylamino)xanthen-10-ium-9-yl]benzoyl}-1,5-diazocan-1-yl)-28-oxoolean-12-ene Chloride (19)
Following GP 3 from 14 (150 mg, 0.14 mmol) and rhodamine B (100 mg, 0.2 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 10% → 40%), 19 (100 mg, 72%) was obtained as a pink solid; m.p. = 211–216 °C; Rf = 0.44 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 562 nm (4.53); IR (ATR): ν = 2926m, 2605w, 2498w, 1729w, 1587s, 1466s, 1412s, 1336, 1245s, 1180s, 1132m, 1073s, 1009m, 921w, 748m, 683m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.67–7.57 (m, 2H, 43-H, 44-H), 7.53–7.47 (m, 1H, 42-H), 7.34–7.26 (m, 3H, 45-H, 48-H), 7.14–6.65 (m, 4H, 49-H, 51-H), 5.25–5.15 (m, 1H, 12-H), 4.51–4.40 (m, 1H, 3-H), 3.78–3.20 (m, 16H, 33-H, 35-H, 36-H, 38-H, 53-H), 3.05–2.95 (m, 1H, 18-H), 2.07–2.02 (m, 1H, 16-Ha), 2.01–1.99 (m, 3H, 32-H), 1.89–1.81 (m, 2H, 11-H), 1.67–1.37 (m, 14H, 19-Ha, 1-Ha, 2-H, 9-H, 6-Ha, 15-Ha, 22-Ha, 21-Ha, 6-Hb, 34-H, 37-H), 1.30–1.24 (m, 12H, 54-H), 1.23–1.13 (m, 6H, 16-Hb, 7-H 22-Hb, 21-Hb, 19-Hb,), 1.08 (s, 3H, 27-H), 0.99 (m, 2H, 1-Hb, 15-Hb), 0.87 (s, 3H, 25-H), 0.86 (s, 3H, 29-H), 0.84 (s, 3H, 30-H), 0.82 (s, 3H, 23-H), 0.80 (s, 3H, 24-H), 0.79–0.76 (m, 1H, 5-H), 0.68 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 170.9 (C-28, C-31), 168.7 (C-39), 157.7 (C-52), 155.8 (C-46), 155.7 (C-50), 145.4 (C-13), 136.6 (C-41), 132.5 (C-49), 130.4 (C-40), 130.1 (C-42), 130.0 (C-44), 129.4 (C-43), 127.7 (C-45), 121.2 (C-129, 113.9 (C-47), 96.1 (C-48, C-51), 80.9 (C-3), 55.3 (C-5), 48.4 (C-17), 47.6 (C-9), 46.6 (C-19), 46.2 (C-53), 46.1 (C-33, C-35, C-36, C-38), 44.7 (C-18), 42.0 (C-14), 39.1 (C-8), 38.0 (C-1), 37.6 (C-4), 37.0 (C-10), 34.1 (C-21), 32.9 (C-30), 32.8 (C-22), 30.3 (C-20), 29.6 (C-7), 28.0 (C-15), 28.0 (C-23), 25.8 (C-27), 24.0 (C-29), 23.5 (C-2), 23.3 (C-11), 22.6 (C-16), 21.3 (C-32), 18.2 (C-6), 17.2 (C-26), 16.6 (C-24), 15.4 (C-25), 12.7 (C-54) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1021.4 (98%, [M-Cl]+); analysis calcd. for C66H91N4O5Cl (1055.93): C 75.07, H 8.69, N 5.31; found: C 74.87, H 8.82, N 5.08.
4.15. (3ß)-3-Acetyloxy-28-(5-{2-[3,6-bis(diethylamino)xanthen-10-ium-9-yl]benzoyl}-1,5-diazocan-1-yl)-28-oxours-12-ene Chloride (20)
Following GP 3 from 15 (150 mg, 0.14 mmol) and rhodamine B (100 mg, 0.2 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 10% → 40%), 20 (94 mg, 63%) was obtained as a pink solid; m.p. = 194–197 °C (decomp.); Rf = 0.41 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 560 nm (5.54); IR (ATR): ν = 2932w, 1726w, 1586s, 1465m, 1411s, 1335s, 1272m, 1245s, 1179s, 1132m, 1073m, 1009m, 921m, 823w, 746m, 683m, 663m, 498m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.68–7.56 (m, 2H, 43-H, 44-H), 7.53–7.49 (m, 1H, 42-H), 7.32–7.25 (m, 3H, 45-H, 48-H), 7.18–6.54 (m, 4H, 49-H, 51-H), 5.24–5.11 (m, 1H, 12-H), 4.50–4.39 (m, 1H, 3-H), 4.12–2.76 (m, 16H, 33-H, 35-H, 36-H, 38-H,53-H), 2.43–2.33 (m, 1H, 18-H), 2.09–2.07 (m, 1H, 16-Ha), 2.00 (s, 3H, 32-H), 1.91–1.84 (m, 2H, 11-H), 1.77–1.36 (m, 14H, 1-Ha, 2-H, 21-Ha, 6-Ha, 9-H, 22-Ha, 19-H, 6-Hb, 16-Hb, 34-H, 37-H), 1.29 (t, J = 7.1 Hz, 12H, 54-H), 1.23 (m, 6H, 7-H, 15-H, 21-Hb, 22-Hb), 1.03 (s, 4H, 1-Hb, 27-H), 0.96–0.93 (m, 1H, 20-H), 0.90 (s, 3H, 29-H), 0.88 (s, 3H, 25-H), 0.83 (s, 3H, 30-H), 0.82 (s, 3H, 23-H), 0.81 (s, 3H, 24-H), 0.77–0.75 (m, 1H, 5-H), 0.69 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 170.9 (C-28), 157.7 (C-31), 157.7 (C-39), 155.8 (C-50), 155.7 (C-46) 155.6 (C-52), 136.6 (C-40), 132.9 (C-48), 130.1 (C-44), 130.0 (C-42), 129.4 (C-43), 127.0 (C-45), 125.0 (C-12), 113.9 (C-47), 96.2 (C-49, C-51), 80.9 (C-3), 55.3 (C-18), 55.3 (C-5), 49.4 (C-17), 47.5 (C-9), 46.2 (C-33, C-35, C-36, C-38), 46.1 (C-53), 42.4 (C-14), 39.6 (C-19), 39.3 (C-8), 38.6 (C-20), 38.2 (C-1), 37.6 (C-4), 36.9 (C-10), 32.7 (C-22), 31.9 (C-7), 30.5 (C-21), 29.6 (C-15), 29.3 (C-16), 28.0 (C-23), 23.2 (C-2), 23.1 (C-11), 23.0 (C-27), 18.1 (C-6), 17.4 (C-30), 17.2 (C-26), 16.7 (C-24), 15.5 (C-25), 12.7 (C-54) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1020.4 (100%, [M-Cl]+); analysis calcd. For C66H91N4O5Cl (1055.93): C 75.07, H 8.69, N 5.31; found: C 74.83, H 8.91, N 5.03.
4.16. (3ß)-3-Acetyloxy-28-(5-{2-[3,6-bis(diethylamino)xanthen-10-ium-9-yl]benzoyl}-1,5-diazocan-1-yl)-28-oxolup-20(29)-ene Chloride (21)
Following GP 3 from 16 (300 mg, 0.5 mmol) and rhodamine B (200 mg, 0.4 mmol), followed by chromatography (silica gel, CHCl3/MeOH, 9:1), 21 (3536 mg, 69%) was obtained as a pink solid; m.p. = 212–218 °C; Rf = 0.49 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 562 nm (4.43); IR (ATR): ν = 2936w, 1730w, 1587s, 1465m, 1411s, 1335s, 1244s, 1179s, 1132m, 1073m, 978w, 921w, 684m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.52–7.47 (m, 2H, 43-H, 44-H), 7.43–7.37 (m, 1H, 42-H), 7.22–6.99 (m, 3H, 45-H, 48-H), 6.97–6.71 (m, 2H, 49-H), 6.64–6.57 (m, 2H, 51-H), 4.58–4.50 (m, 1H, 29-Ha), 4.42–4.37 (m, 1H, 29-Hb), 4.30 (m, 1H, 3-H), 3.73–2.93 (m, 16H, 33-H, 35-H, 36-H, 38-H, 53-H), 2.83–2.68 (m, 2H, 19-H, 13-H), 2.00–1.94 (m, 1H, 16-Ha), 1.87 (s, 3H, 32-H), 1.83–1.54 (m, 4H, 22-Ha, 15-H, 21-Ha), 1.53–1.51 (m, 2H, 12-Ha, 1-Ha), 1.50 (s, 3H, 30-H), 1.47–1.42 (m, 2H, 2-H), 1.41–1.36 (m, 1H, 18-H), 1.35–1.29 (m, 2H, 16-Ha, 6-Ha), 1.28–1.12 (m, 21H, 11-Ha, 6-Hb, 7-H, 22-Hb, 54-H, 34-H, 37-H), 1.11–1.09 (m, 2H, 11-Hb, 9-H, 21-Hb), 0.83–0.74 (m, 8H, 1-Hb, 12-Hb, 23-H, 27-H), 0.69–0.65 (m, 9H, 26-H, 25-H, 24-H), 0.63–0.59 (m, 1H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 170.8 (C-28), 168.5 (C-39), 167.6 (C-31), 157.6 (C-50), 155.6 (C-46), 155.5 (C-52), 151.2 (C-20), 136.4 (C-40), 132.3 (C-41), 130.8 (C-48), 130.0 (C-43), 129.4 (C-42), 128.6 (C-45), 127.0 (C-44), 114.6 (C-49), 113.6 (C-47), 108.9 (C-29), 96.0 (C-51), 80.8 (C-3), 55.2 (C-5), 55.1 (C-38, C-36, C-35, C-33), 53.0 (C-18), 50.6 (C-9), 46.1 (C-53), 45.8 (C-19), 41.9 (C-17), 40.6 (C-8), 40.6 (C-14), 38.3 (C-1), 37.7 (C-10), 37.0 (C-4), 36.8 (C-13), 36.0 (C-22), 34.2 (C-7), 32.0 (C-16), 31.3 (C-21), 30.2 (C-34, C-37), 29.8 (C-15), 27.8 (C-24), 25.5 (C-12), 25.4 (C-37, C-34), 23.6 (C-2), 21.2 (C-32), 21.0 (C-11), 19.6 (C-30), 18.1 (C-6), 16.1 (C-25), 15.9 (C-26), 14.6 (C-23), 14.5 (C-27), 12.6 (C-54) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1020.5 (100%, [M-Cl]+); analysis calcd. for C66H91N4O5Cl (1055.93): C 75.07, H 8.69, N 5.31; found: C 74.86, H 8.90, N 5.09.
4.17. (3ß)-3-Acetyloxy-28-(5-{2-[3,6-bis(diethylamino)xanthen-10-ium-9-yl]benzoyl}-1,5-diazocan-1-yl)-28-oxolup-20-oxo Chloride (22)
Following GP 3 from 17 (300 mg, 0.50 mmol) and rhodamine B (300 mg, 0.6 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 9:1), 22 (350 mg, 69%) was obtained as a pink solid; m.p. = 198–201 °C; Rf = 0.51 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 558 nm (4.73); IR (ATR): ν = 2934w, 1721m, 1585s, 1410m, 1466s, 1334s, 1272s, 1245s, 1131s, 1072s, 1009s, 977m, 921m, 823m, 755m, 682s cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.67–7.57 (m, 2H, 42-H, 43-H), 7.53–7.47 (m, 1H, 41-H), 7.35–7.27 (m, 3H, 44-H, 47-H), 7.11–6.64 (m, 4H, 48-H, 50-H), 4.46–4.36 (m, 1H, 3-H), 3.88–3.20 (m, 16H, 32-H, 34-H, 35-H, 37-H, 52-H), 3.16–3.05 (m, 1H, 18-H), 2.76–2.51 (m, 1H, 13-H), 2.11–2.04 (m, 4H, 16-Ha, 29-H), 1.98 (s, 4H, 19-H, 31-H), 1.78 (s, 2H, 21-Ha, 22-Ha), 1.62–1.48 (m, 4H, 1-Ha, 2-H, 16-Ha), 1.42 (m, 7H, 6-Ha, 22-Hb, 21-Hb, 11-H, 7-Ha, 6-Hb), 1.28 (t, J = 6.8 Hz, 14H, 7-Hb, 9-H, 53-H), 1.23–1.04 (m, 6H, 33-H, 36-H, 15-H), 0.94 (s, 2H, 12-H), 0.91 (s, 4H, 1-Hb, 24-H), 0.82 (s, 3H, 27-H), 0.78 (m, 10H, 23-H, 25-H, 26-H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 212.8 (C-20), 170.9 (C-28, C-30, C- 38), 157.7 (C-49), 155.7 (C-45), 155.6 (C-51), 136.6 (C-39), 136.5 (C-40), 132.4 (C-47), 130.1 (C-42), 129.7 (C-43), 129.4 (C-44), 127.1 (C-41), 114.5 (C-48), 113.7 (C-46), 96.2 (C-50), 80.8 (C-3), 55.4 (C-5), 55.3 (C-32, C-34, C-35, C-37), 53.0 (C-19), 50.6 (C-9), 50.3 (C-18), 49.5 (C-17), 46.2 (C-52), 41.9 (C-14), 40.6 (C-8), 38.3 (C-1), 37.7 (C-4), 37.1 (C-10), 35.8 (C-13), 35.6 (C-22), 34.2 (C-7), 31.6 (C-16), 30.1 (C-29), 29.9 (C-15), 28.8 (C-21), 27.9 (C-23), 27.4 (C-12), 23.6 (C-2), 22.6 (C-33, C-36), 21.1 (C-11), 18.1 (C-6), 16.4 (C-25), 16.2 (C-26), 14.7 (C-24), 14.0 (C-27), 12.7 (C-53) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1022.4 (100%, [M-Cl]+); analysis calcd. for C65H89N4O6Cl (1057.90): C 73.80, H 8.48, N 5.30; found: C 73.55, H 8.67, N 5.07.
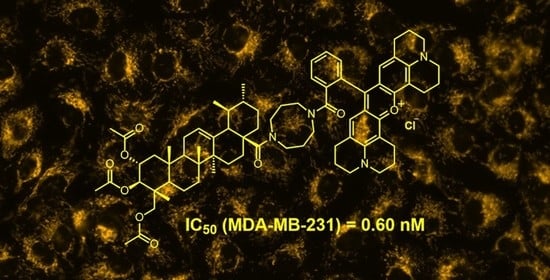
4.18. (2α,3ß,4α)2,3,23-Tris (acetyloxy)-28-(5-{2-[3,6-bis(diethylamino)xanthen-10-ium-9-yl]benzoyl}-1,5-diazocan-1-yl)-28-oxours-12-en Chloride (23)
Following GP 3 from 18 (300 mg, 0.4 mmol) and rhodamine B (250 mg, 0.5 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 9:1), 23 (184 mg, 60%) was obtained as a pink solid; m.p. = 225 °C; Rf = 0.44 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 562 nm (4.50); IR (ATR): ν = 2927w, 1793m, 1587s, 1467m, 1411m, 1336s, 1244s, 1179s, 1042m, 921w, 684m, 436w cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.66–7.57 (m, 2H, 47-H, 48-H), 7.52–7.47 (m, 1H, 46-H), 7.27-7.20 (m, 49-H, 52-H), 7.17–6.63 (m, 4H, 53-H, 55-H), 5.20–5.00 (m, 3H, 12-H, 2-H, 3-H), 3.82–3.76 (m, 1H, 23-Ha), 3.73–2.93 (m, 17H, 37-H, 39-H, 40-H, 42-H, 57-H, 23-Hb), 2.44–2.33 (m, 1H, 18-H), 2.04 (s, 3H, 34-H), 2.02–1.99 (m, 1H, 1-Ha), 1.97 (s, 3H, 36-H), 1.93 (s, 3H, 32-H), 1.90–1.32 (m, 15H, 11-H, 9-H, 15-H, 16-Ha, 21-Ha, 22-Ha, 20-H, 38-H, 41-H, 6-H), 1.28 (t, J = 7.1 Hz, 13H, 5-H, 58-H), 1.25–1.10 (m, 5H, 7-H, 16-Hb, 21-Hb, 22-Hb), 1.09–1.07 (m, 1H, 1-Hb), 1.04 (s, 3H, 30-H), 1.01 (s, 3H, 27-H), 0.97–0.92 (m, 1H, 19-H), 0.88 (s, 3H, 29-H), 0.84 (s, 3H, 25-H), 0.81 (s, 3H, 26-H), 0.69 (s, 3H, 24-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 176.2 (C-43), 170.8 (C-35), 170.4 (C-31, C-33), 170.3 (C-28), 157.7 (C-54), 155.7 (C-56), 155.6 (C-50), 138.6 (C-13), 136.6 (C-45), 132.3 (C-52), 130.1 (C-47), 130.0 (C-49), 129.3 (C-48), 127.2 (C-46), 125.9 (C-12), 113.9 (C-51), 96.2 (C-53, C-55), 74.8 (C-3), 69.9 (C-2), 65.3 (C-23), 55.5 (C-18), 53.4 (C-37, C-39, C-40, C-42), 47.6 (C-5), 47.5 (C-9), 46.2 (C-57), 46.1 (C-17), 43.7 (C-1), 42.5 (C-4), 41.9 (C-14), 38.9 (C-8), 38.7 (C-20), 38.6 (C-19), 37.8 (C-10), 32.6 (C-22), 30.5 (C-21), 29.6 (C-7), 28.4 (C-15), 23.4 (C-27), 23.3 (C-11), 21.2 (C-29), 21.0 (C-32), 20.8 (C-36), 20.7 (C-34), 17.8 (C-6), 17.4 (C-26), 17.2 (C-30), 17.0 (C-24), 13.9 (C-25), 12.6 (C-58) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1036.5 (100%, [M-Cl]+); analysis calcd. for C70H95N4O9Cl (1172.00): C 71.74, H 8.17, N 4.78; found: C 71.49, H 8.35, N 4.47.
4.19. 3β-Acetyloxy-28-[4-[3-(2,3,6,7,12,13,16,17-octahydro-1H,5H,11H,15H-pyrido [3,2,1-ij]pyrido[1’’,2’’,3’’:1’,8’]quinolino[6’,5’:5,6]pyrano[2,3-f ]quinolin-4-ium-9-yl)benzoyl]1,5-diazocan-1-yl]-28-oxo-olean-12-en Chloride (24)
Following GP 3 from 14 (100 mg, 0.14 mmol) and rhodamine 101 (200 mg, 0.4 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 10% → 50%), 24 (114 mg, 75%) was obtained as a pink solid; m.p. = 205–210 °C; Rf = 0.41 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 580 nm (4.23); IR (ATR): ν = 2942w, 1727m, 1595s, 1493m, 1459m, 1362m, 1295s, 1196s, 1035m, 746m, 420m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.81–7.61 (m, 2H, 45-H), 7.54–7.43 (m, 1H, 42-H), 7.29 (s, 1H, 44-H), 6.86–6.47 (m, 2H, 48-H), 5.27–5.21 (m, 1H, 12-H), 4.51–4.44 (m, 1H, 3-H), 3.79–3.15 (m, 16H, 33-H, 35-H, 36-H, 38-H, 52-H, 57-H), 3.09–2.90 (m, 5H, 18-H, 55-H,), 2.76–2.47 (m, 4H, 50-H), 2.15–2.06 (m, 4H, 56-H), 2.03 (s, 3H, 32-H), 1.97 (s, 5H, 16-Ha, 51-H), 1.85 (s, 2H, 11-H), 1.70–1.15 (m, 20H, 19-Ha, 21-H, 2-H, 1-Ha, 9-H, 6-Ha, 7-Ha, 6-Hb, 22-Ha, 7-Hb, 15-H, 22-Hb, 19-Hb, 34-H, 37-H), 1.12 (s, 3H, 30-H), 1.07–0.97 (m, 2H, 1-Hb, 16-Hb), 0.91 (s, 3H, 25-H), 0.90 (s, 3H, 27-H), 0.88 (s, 3H, 29-H), 0.85 (s, 3H, 23-H), 0.83 (s, 3H, 24-H), 0.81–0.79 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.0 (C-28, C-31, C-39), 164.1 (C-53), 152.0 (C-46), 139.9 (C-58), 134.6 (C-41), 131.0 (C-40), 130.5 (C-44), 129.4 (C-45, C-42), 126.9 (C-43), 125.9 (C-48), 123.5 (C-47), 121.4 (C-12), 113.0 (C-49), 105.3 (C-54), 81.0 (C-3), 55.4 (C-5), 51.1 (C-33, C-35, C-36, C-38), 50.5 (C-52, C-57), 48.2 (C-17), 47.6 (C-9), 46.6 (C-19), 43.7 (C-18), 43.3 (C-14), 39.1 (C-8), 38.1 (C-1), 37.7 (C-4), 37.0 (C-10), 33.9 (C-22), 33.0 (C-29), 32.9 (C-7), 30.5 (C-20), 30.3 (C-21), 29.7 (C-15), 28.0 (C-23), 27.8 (C-16), 27.6 (C-50), 25.8 (C-30), 24.1 (C-27), 23.5 (C-2), 23.4 (C-11), 21.3 (C-32), 20.6 (C-51), 19.9 (C-55), 19.7 (C-56), 18.2 (C-6), 17.2 (C-26), 16.7 (C-24), 15.4 (C-25) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1068.6 (100%, [M-Cl]+); analysis calcd. For C70H91N4O5Cl (1103.97): C 76.16, H 8.31, N 5.08; found: C 75.81, H 8.52, N 4.89.
4.20. 3β-Acetyloxy-28-[4-[3-(2,3,6,7,12,13,16,17-octahydro-1H,5H,11H,15H-pyrido[3,2,1-ij]pyrido[1’’,2’’,3’’:1’,8’]quinolino[6’,5’:5,6]pyrano[2,3-f]uinoline-4-ium-9-yl)benzoyl]1,5-diazocan-1-yl]-28-oxo-urs-12-en Chloride (25)
Following GP 3 from 15 (150 mg, 0.2 mmol) and rhodamine 101 (150 mg, 0.3 mmol), followed by chromatography (silica gel, ethyl acetate/MeOH, 10% → 50%), 25 (94 mg, 62%) was obtained as a pink solid; m.p. = 199–202 °C; Rf = 0.43 (CHCl3:Methanol, 9:1); UV-Vis (CHCl3): λmax (log ε) = 571 nm (3.94); IR (ATR): ν = 3388w, 2925m, 1728m, 1597s, 1495m, 1459m, 1362s, 1297s, 1246s, 1195s, 1100s, 1024s, 421s cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.36–8.06 (m, 1H, 43-H), 7.75–7.63 (m, 2H, 42-H, 45-H), 7.24–7.11 (m, 1H, 44-H), 6.81–6.50 (m, 2H, 48-H), 5.26–5.16 (m, 1H, 12-H), 4.49–4.43 (m, 1H, 3-H), 3.71–3.21 (m, 16H, 33-H, 35-H, 36-H, 38-H, 52-H, 57-H), 3.17–2.90 (m, 4H, 55-H), 2.81–2.57 (m, 4H, 50-H), 2.23–2.06 (m, 4H, 56-H), 2.01 (s, 3H, 32-H), 1.98–1.84 (m, 6H, 51-H, 11-Ha, 16-Ha), 1.66–1.19 (m, 22H, 1-Ha, 11-Hb, 21-Ha, 6-Ha, 22-Ha, 19-H, 6-Hb, 21-Hb, 22-Hb, 2-H, 15-H, 7-H, 16-Hb, 18-H, 34-H, 37-H), 1.13–1.10 (m, 3H, 29-H), 1.05 (s, 3H, 27-H), 1.04–0.99 (m, 2H, 1-Hb, 20-H), 0.92 (s, 3H, 24-H), 0.85 (s, 3H, 25-H), 0.83 (s, 3H, 23-H), 0.82 (s, 3H, 30-H), 0.79–0.77 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.0 (C-28), 169.3 (C-31, C-39), 151.2 (C-53), 150.9 (C-46), 135.2 (C-58), 132.3 (C-42), 131.3 (C-43), 130.2 (C-45), 129.1 (C-44), 127.1 (C-48), 125.1 (C-12), 112.6 (C-49), 111.6 (C-47), 105.6 (C-54), 80.9 (C-3), 55.3 (C-5), 47.7 (C-33, C-35, C-36, C-38), 47.5 (C-18), 47.5 (C-9), 45.3 (C-17), 43.3 (C-52, C-57), 41.7 (C-14), 39.6 (C-8), 39.5 (C-19), 38.6 (C-20), 38.2 (C-1), 37.7 (C-4), 36.9 (C-10), 33.1 (C-22), 31.9 (C-7), 30.5 (C-21), 29.7 (C-15), 28.0 (C-16), 27.8 (C-23), 27.5 (C-50), 25.0 (C-34, C-37), 23.5 (C-11), 23.4 (C-27), 23.3 (C-51), 22.6 (C-2), 21.3 (C-32), 19.9 (C-55), 19.7 (C-56), 18.7 (C-29), 18.1 (C-6), 17.3 (C-30), 16.7 (C-26), 15.5 (C-24), 14.1 (C-25) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1068.4 (100%, [M-Cl]+); analysis calcd. for C70H91N4O5Cl (1103.97): C 76.16, H 8.31, N 5.08; found: C 75.87, H 8.59, N 4.83.
4.21. 3β-Acetyloxy-28-[4-[3-(2,3,6,7,12,13,16,17-octahydro-1H,5H,11H,15H-pyrido[3,2,1-ij] pyrido[1”,2”,3”:1’,8’]quinolino[6’,5’:5,6]pyrano[2,3-f]quinolin-4-ium-9-yl)benzoyl] 1,5-diazocan-1-yl]-28-oxo-lup-20(29)-en Chloride (26)
Following GP 3 from 16 (200 mg, 0.14 mmol) and rhodamine 101 (200 mg, 0.4 mmol), followed by chromatography (silica gel, CHCl3/MeOH, 9:1), 26 (103 mg, 68%) was obtained as a pink solid; m.p. = 203–206 °C; Rf = 0.44 (CHCl3/MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 578 nm (4.33); IR (ATR): ν = 2931w, 1721w, 1595s, 1493s, 1361m, 1294s, 1246s, 1180s, 1035s, 746m, 622m, 421s cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.68–7.53 (m, 2H, 43-H, 45-H), 7.52–7.45 (m, 1H, 42-H), 7.31–7.26 (m, 1H, 44-H), 6.76–6.59 (m, 2H, 48-H), 4.70–4.62 (m, 1H, 29-Ha), 4.54–4.49 (m, 1H, 29-Hb), 4.44–4.38 (m, 1H, 3-H), 3.81–3.02 (m, 16H, 33-H, 35-H, 36-H, 38-H, 52-H, 57-H), 3.00–2.89 (m, 4H, 55-H), 2.88–2.72 (m, 2H, 13-H, 18-H), 2.71–2.51 (m, 4H, 50-H), 2.16–2.01 (m, 5H, 16-Ha, 56-H), 1.99 (s, 3H, 32-H), 1.96–1.87 (m, 5H, 21-Ha, 51-H), 1.85–1.74 (m, 2H, 15-Ha, 22-Ha), 1.70–1.66 (m, 1H, 12-Ha), 1.62 (s, 4H, 1-Ha, 30-H), 1.60–1.53 (m, 2H, 2-H), 1.50–1.47 (m, 1H, 9-H), 1.47–1.40 (m, 2H, 6-Ha, 16-Hb), 1.36–1.06 (m, 13H, 11-Ha, 21-Hb, 6-Hb, 7-H, 15-Hb, 19-H, 22-Hb, 11-Hb, 34-H, 37-H), 0.94 (s, 2H, 1-Hb, 12-Hb), 0.90 (s, 3H, 24-H), 0.86 (s, 3H, 25-H), 0.81 (s, 3H, 27-H), 0.79 (s, 3H, 23-H), 0.73 (s, 3H, 26-H), 0.70–0.59 (m, 1H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 170.9 (C-28), 168.8 (C-31, C-39), 151.9 (C-53), 151.3 (C-46), 151.2 (C-20), 138.4 (C-40, C-41), 136.5 (C-58), 130.4 (C-44), 129.4 (C-45, C-42), 127.1 (C-48), 127.0 (C-43) 123.5 (C-47), 113.1 (C-49), 109.1 (C-29), 105.3 (C-54), 81.0 (C-3), 55.5 (C-5), 53.1 (C-9), 52.9, 50.9 (C-33, C-35, C-36, C-38), 50.7 (C-19), 50.5 (C-52, C-57), 49.4 (C-17), 45.9 (C-13), 42.0 (C-14), 40.7 (C-14), 40.6 (C-8), 38.4 (C-1), 37.8 (C-4), 37.1 (C-10), 36.9 (C-18), 36.1 (C-21), 34.3 (C-7), 32.1 (C-16), 31.4 (C-22), 29.9 (C-15), 27.9 (C-23), 27.5 (C-50), 25.5 (C-12), 23.7 (C-2), 22.6 (C-34, C-37), 21.3 (C-32), 21.1 (C-11), 20.6 (C-51), 19.8 (C-55), 19.6 (C-56), 18.7 (C-30), 18.2 (C-6), 16.5 (C-25), 16.4 (C-26), 14.7 (C-24), 14.6 (C-27) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1067.2 (100%, [M-Cl]+); analysis calcd. for C70H91N4O5Cl (1103.97): C 76.16, H 8.31, N 5.08; found: C 75.98, H 8.52, N 4.83.
4.22. 3β-Acetyloxy-28-[4-[3-(2,3,6,7,12,13,16,17-octahydro-1H,5H,11H,15H-pyrido[3,2,1-ij] pyrido[1”,2”,3”:1’,8’]quinolino[6’,5’:5,6]pyrano[2,3-f]quinolin-4-ium-9-yl)benzoyl] 1,5-diazocan-1-yl]-30-nor-20,28-dioxo-lup-20(29)-en Chloride (27)
Following GP 3 from 17 (200 mg, 0.3 mmol) and rhodamine 101 (100 mg, 0.2 mmol), followed by chromatography (silica gel, CHCl3/MeOH, 9:1), 27 (132 mg, 60%) was obtained as a pink solid; m.p. = 208–210 °C; Rf = 0.49 (CHCl3:MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 577 nm (4.66); IR (ATR): ν = 3350w, 1596s, 1195s, 1298s, 1197s, 1138s cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.63–7.52 (m, 2H, 41-H, 44-H), 7.50–7.43 (m, 1H, 42-H), 7.24–7.20 (m, 1H, 43-H), 6.72–6.59 (m, 2H, 47-H), 4.42–4.33 (m, 1H, 3-H), 3.86–3.01 (m, 17H, 18-H, 32-H, 34-H, 35-H, 37H, 49-H, 51-H), 2.97–2.85 (m, 4H, 54-H), 2.76–2.54 (m, 4H, 49-H), 2.52–2.43 (m, 1H, 13-H), 2.18–2.06 (m, 4H, 21-Hb, 29-H), 2.07–1.99 (m, 4H, 55-H), 1.96 (s, 4H, 19-H, 31-H), 1.94–1.64 (m, 7H, 50-H, 22-Ha, 16-Ha, 15-Ha), 1.60–1.14 (m, 18H, 1-Ha, 2-H, 21-Hb, 22-Hb, 6-Ha, 16-Hb, 11-Ha, 7-H, 6-Hb, 11-Hb, 15-Hb), 1.08–0.96 (m, 3H, 1-Hb, 12-H), 0.90 (s, 3H, 24-H), 0.85 (s, 3H, 25-H), 0.80 (s, 3H, 27-H), 0.76 (s, 3H, 23-H), 0.71 (s, 3H, 26-H), 0.69–0.65 (m, 1H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 213.2 (C-20), 170.9 (C-28, C-30, C-38), 151.9 (C-52), 151.2 (C-45), 136.6 (C-57), 136.3 (C-39, C-40), 130.4 (C-43), 129.4 (C-41, C-44), 126.8 (C-42), 126.7 (C-47), 123.5 (C-46), 113.0 (C-48), 105.2 (C-53), 80.8 (C-3), 55.4 (C-5), 53.0 (C-19), 50.9 (C-32, C-34, C-35, C-37), 50.6 (C-9), 50.4 (C-51, C-56), 50.3 (C-18), 49.8 (C-17), 44.1 (C-13), 41.8 (C-14), 40.6 (C-8), 38.3 (C-1), 37.7 (C-10), 37.1 (C-4), 35.8 (C-22), 34.2 (C-7), 31.8 (C-21), 30.1 (C-29), 29.9 (C-15), 28.8 (C-16), 27.9 (C-23), 27.5 (C-49), 27.3 (C-12), 23.6 (C-2), 22.6 (C-33, C-36), 21.2 (C-31), 21.1 (C-11), 20.6 (C-50), 19.9 (C-54), 19.6 (C-55), 18.1 (C-6), 16.4 (C-26), 15.9 (C-25), 14.7 (C-24), 14.0 (C-27) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1070 (100%, [M-Cl]+); analysis calcd. for C69H89N4O6Cl (1105.94): C 74.94, H 8.11, N 5.07; found: C 74.73, H 8.35, N 4.81.
4.23. (2α,3ß,4α)2,3,23-Tris(acetoxy)-28-[4-[3-(2,3,6,7,12,13,16,17-octahydro-1H,5H,11H,15H-pyrido[3,2,1-ij]pyrido[1’’,2’’,3’’:1’,8’]quinolino[6’,5’:5,6]pyrano[2,3-f]quinolin-4-ium-9-yl)benzoyl]1,5-diazocan-1-yl]-28-oxo-olean-12-en Chloride (28)
Following GP 3 from 18 (200 mg, 0.3 mmol) and rhodamine 101 (200 mg, 0.4 mmol), followed by chromatography (silica gel, CHCl3/MeOH, 9:1), 28 (232 mg, 64%) was obtained as a pink solid; m.p. = 193–196 °C; Rf = 0.45 (CHCl3:MeOH, 9:1); UV-Vis (CHCl3): λmax (log ε) = 578 nm (4.50); IR (ATR): ν = 2924w, 1739w, 1594s, 1493s, 1459m, 1361m, 1293s, 1195s, 1180s, 1090s, 1035s, 729m, 622m, 421s cm−1; 1H NMR (400 MHz, CDCl3): δ =7.67–7.58 (m, 2H, 47-H, 49-H), 7.52–7.48 (m, 1H, 46-H), 7.29–7.27 (m, 1H, 48-H), 6.77–6.65 (m, 2H, 52-H), 5.17–5.03 (m, 3H, 12-H, 2-H, 3-H), 3.83–3.79 (m, 1H, 23-Ha), 3.60–3.16 (m, 17H, 23-Hb, 37-H, 39-H, 40-H, 42-H, 56-H, 61-H), 3.00–2.94 (m, 4H, 59-H), 2.77–2.63 (m, 4H, 54-H), 2.46–2.36 (m, 1H, 18-H), 2.07 (s, 4H, 60-H), 2.06 (s, 3H, 36-H), 2.04–2.01 (m, 1H, 1-Ha), 1.99 (s, 3H, 34-H), 1.95 (s, 7H, 32-H, 55-H), 1.91–1.86 (m, 2H, 11-H), 1.60–1.57 (m, 1H, 9-H), 1.46–1.42 (m, 2H, 21-Ha, 22-Hb), 1.35–1.30 (m, 4H, 6-H, 19-H, 5-H), 1.26–1.22 (m, 12H, 16-Ha, 38-H, 41-H, 22-Hb, 7-H, 15-H, 21-Hb, 16-Hb), 1.11–1.09 (m, 1H, 1-Hb), 1.04 (s, 3H, 24-H), 0.98–0.94 (m, 1H, 20-H), 0.91 (s, 3H, 29-H), 0.85 (s, 3H, 25-H), 0.83 (s, 3H, 27-H), 0.82 (s, 3H, 30-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 176.3 (C-28), 170.8 (C-35, C-43), 170.4 (C-33), 170.3 (C-31), 152.0 (C-57), 151.3 (C-50), 139.1 (C-62), 136.6 (C-44), 130.3 (C-45), 129.6 (C-48), 129.2 (C-46) 129.1 (C-49), 127.0 (C-47, C-52), 124.5 (C-12), 123.4 (C-51), 113.0 (C-53), 105.2 (C-58), 74.9 (C-3), 69.9 (C-2), 65.3 (C-23), 55.6 (C-18), 51.0 (C-34, C-37, C-40, C-42), 50.5 (C-56, C-61), 47.7 (C-5), 47.5 (C-9), 46.2 (C-17), 43.7 (C-1), 41.9 (C-4, C-14), 39.5 (C-19), 38.6 (C-20), 37.8 (C-10), 32.6 (C-22), 31.9 (C-7), 30.6 (C-21), 29.7 (C-15), 29.6 (C-16), 27.6 (C-54), 23.3 (C-11), 22.6 (C-38, C-41), 22.6 (C-27), 21.2 (C-29), 21.0 (C-32), 20.8 (C-36), 20.7 (C-34), 20.6 (C-55), 19.9 (C-59), 19.7 (C-60), 17.9 (C-6), 17.4 (C-30), 17.1 (C-24), 17.0 (C-26), 14.1 (C-25) ppm; MS (ESI, MeOH/CHCl3, 4:1): m/z = 1084.3 (100%, [M-Cl]+); analysis calcd. for C74H95N4O9Cl (1220.04): C 72.85, H 7.85, N 4.59; found: C 72.63, H 8.01, N 4.39.
4.24. Cell Culture
Breast cancer cell lines were obtained from the Department of Radiobiology (MLU Halle-Wittenberg) and previously described. MDA-MB-231, HS578T and MCF-7, and T47D were cultured as a monolayer in RPMI (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (Capricorn Scientific, Ebsdorfergrund, Germany), 2% penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA), and 1% sodium pyruvate (Gibco, Thermo Fisher Scientific) at 37 °C and 5% CO2. All cell lines were regularly tested for mycoplasma contamination.
4.25. SRB Assay
Breast cancer cells were seeded in 96 well plates with different cell numbers depending on the cell line in triplicate and after 24 h treated with different concentrations of compounds 14–28. Treatment ended after 96 h when cells were fixed with 10% trichloroacetic acid (Carl Roth GmbH, Karlsruhe, Germany) for 1h at 4 °C. Afterwards, cells were washed with ice water four times and stained with 4.4% SRB solution (Sigma–Aldrich) for 10 min at room temperature. After washing cells with 1% acetic acid (Carl Roth GmbH), cells were air-dried overnight and then dissolved with 300 µL 20 mM Tris base solution (Sigma-Aldrich). Excitation was measured at 540 nm with a Spark plate reader (Tecan Treading AG, Männedorf, Switzerland) and IC50 values were calculated by dose-response curve fitting using Origin 2019 (OriginLab Corp., Northampton, MA, USA).
4.26. Cell Death
For the determination of apoptotic and necrotic cell death after treatment with compound 28, Annexin V-Sytox Deep Red staining was performed. Therefore, MDA-MB-231 and HS578T cells were seeded in 6-well plates. After 24 h, the cells were treated with different concentrations of compound 28 (10 nM, 100 nM, 250 nM, 500 nM, 1 µM, and 2 µM) for 24 h, 48 h, and 72 h at 37 °C and 5% CO2. For analysis of cell death, detached cells were collected in tubes and living cells were detached by accutase (Biowest, Nuaillé, France) and collected in the same tube. After several washing steps cells were resuspended in 1x annexin V binding puffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2) and stained with 5 µL Annexin V-FITC (BioLegend, San Diego, CA, USA) and 1 µL 100 µM Sytox Deep Red Nucleic Acid Stain (Invitrogen, Thermo Fisher Scientific) for 15 min. Afterward, 400 µL 1x annexin V binding puffer were added to each tube. Gating was realized by the use of unstained, single annexin V-FITC or single Sytox Deep Red Nucleic Acid-stained cells, respectively. For quantification of necrotic and apoptotic cells, 10,000 cells were analyzed by LSRFortessa™ flow cytometer (BD Biosciences, Heidelberg, Germany).
4.27. Proliferation
MDA-MB-231 and HS578T cells were seeded in 6-well plates and treated with different concentrations (10 nM, 100 nM, 250 nM, 500 nM, 1 µM, and 2 µM) of compound 28 after 24 h. The number of dead and viable cells was measured by use of a CASY cell counter (OMNI Life Science, Bremen, Germany) after 72 h.
4.28. Staining
Analysis of subcellular localization of compound AS101 was performed in MDA-MB-231 cells using the mitochondrial targeting compound BioTracker™ 488 Green Mitochondria Dye (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) for comparison. Cells were seeded in a µ-Plate 96 Well Black plate (ibiTreat: #1.5 polymer coverslip bottom, ibidi GmbH, Gräfelfing, Germany) at a cell density of 50,000 per well. After 24 h, cells were treated with 100 nM AS101 for 6h or 100 nM BioTracker488 for 30 min, followed by rinsing and supplementation with RPMI 1640 w/o Phenol-red (Pan-Biotech GmbH, Aidenbach, Germany). Live-cell imaging was performed on an Axio Observer 7 (Carl Zeiss Microscopy Deutschland GmbH, Oberkochen, Germany) using the settings for Ex/Em as follows: BioTracker (475 nm/514 nm), AS101 (555 nm/592); Scale bar: 50 µm.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}