
Interplay between Systemic Glycemia and Neuroprotective Activity of Resveratrol in Modulating Astrocyte SIRT1 Response to Neuroinflammation
 , and
, and
Abstract
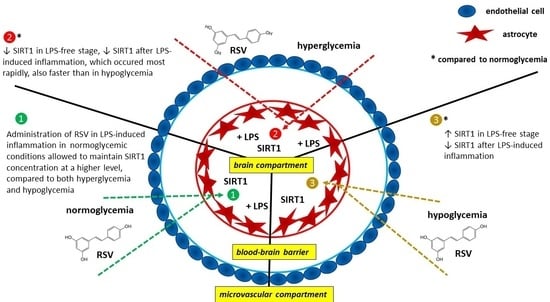
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. SIRT1 Secretion from Astrocytes Depends on Systemic Glycemia
2.2. Neuroinflammation Does Not Alter Astrocyte Morphology and Viability
2.3. Neuroinflammation Hinders Astrocyte SIRT1 Secretion in All Glycemic Backgrounds
2.4. Resveratrol Modulates Basal Astrocyte SIRT1 Secretion Depending on the Systemic Glycemia
2.5. Resveratrol Partially Restores Astrocyte SIRT1 Secretion Hindered by Neuroinflammation, Depending on the Glycemic Background
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Setup of In Vitro Model of the Blood–Brain Barrier (BBB) in Different Glycemic Backgrounds
4.3. Induction of Neuroinflammation
4.4. Administration of Resveratrol (RSV)
4.5. Glucose Colorimetric Assay
4.6. Enzyme Linked Immunosorbent Assay (ELISA)
4.7. Live Cell Fixing and Microscopic Imaging
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nag, S.; Begley, D. Blood Brain Barrier, exchange of metabolites and gases. In Pathology and Genetics of Cerebrovascular Diseases; ISN Neuropath Press: Basel, Switzerland, 2005; pp. 22–29. [Google Scholar]
- Pardridge, W.M. Blood-brain barrier biology and methodology. J. Neurovirol. 1999, 5, 556–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.N. Physiology of the blood-brain barrier and its consequences for drug transport to the brain. Cell. Mol. Neurobiol. 2005, 25, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.N.; Patabendige, A.A.K.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.F.; Leininger-Muller, B.; Cecchelli, R.; Fenstermacher, J.D. Blood-brain interfaces: Relevance to cerebral drug metabolism. Toxicol. Lett. 1995, 82–83, 645–653. [Google Scholar] [CrossRef]
- Shawahna, R.; Uchida, Y.; Decleves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.O.; et al. Transcriptomic and quantitative proteomic analysis of transporters and drug-metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 2011, 8, 1332–1341. [Google Scholar] [CrossRef]
- Ghosh, C.; Puvenna, V.; Gonzalez-Martinez, J.; Jangiro, D.; Marchi, N. Blood-brain barrier P450 enzymes and multidrug transporters in drug resistance: A synergistic role in neurological diseases. Curr. Drug Metab. 2011, 12, 742–749. [Google Scholar] [CrossRef] [Green Version]
- Dauchy, S.; Dutheil, F.; Weaver, R.J.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.O.; Sherrmann, J.M.; De Waziers, I.; Decleves, X. ABC transporters, cytochromes P450 and their main transcription factors: Expression at the human blood-brain barrier. J. Neurochem. 2008, 107, 1518–1528. [Google Scholar] [CrossRef]
- Matsuoka, R.L.; Buck, L.D.; Vajrala, K.P.; Quick, R.E.; Card, O.A. Historical and current perspectives on blood endothelial cell hetero- geneity in the brain. Cell. Mol. Life Sci. 2022, 79, 372. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Wolburg-Buchholz, K.; Mack, A.; Fallier-Becker, P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist 2009, 15, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neuronal circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadao, P.A.C.; Santos, K.B.S.; Vieira, T.H.F.E.; Cordeiro, T.M.E.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef] [PubMed]
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the significance of blood-brain barrier disruption in multiple sclerosis pathology and treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Romanescu, C.; Popescu, B.O. The blood-brain barrier—A key player in multiple sclerosis disease mechanisms. Biomolecules 2022, 12, 538. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. The blood-brain barrier in multiple sclerosis: Functional roles and therapeutic targeting. Autoimmunity 2007, 40, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, S.; Cheng, G.; Guo, B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS ONE 2011, 6, e28432. [Google Scholar] [CrossRef] [Green Version]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Buse, J.B.; Tuomilehto, J.; Fleming, G.A.; Ferrannini, E.; Gerstein, H.C.; Bennett, P.H.; Ramachandran, A. Update and next steps for real-world translation of interventions for type 2 diabetes prevention: Reflections from a diabetes care editors’ expert forum. Diabetes Care 2016, 39, 1186–1201. [Google Scholar] [CrossRef] [Green Version]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, N.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus type 1 and type 2 mouse models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef]
- Rebelos, E.; Rinne, J.O.; Nuutila, P.; Ekblad, L.L. Brain glucose metabolism in health, obesity and cognitive decline—Does insulin have anything to do with it? A narrative review. J. Clin. Med. 2021, 10, 1532. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, L.; Gong, A.; Zhang, J.; Jing, L.; Ding, T.; Li, P.A.; Zhang, J.Z. Damage to the blood-brain barrier and activation of neuroinflammation by focal cerebral ischemia under hyperglycemic condition. Int. J. Mol. Med. 2021, 48, 142. [Google Scholar] [CrossRef] [PubMed]
- Watt, C.; Sanchez-Rangel, E.; Hwang, J.J. Glycemic variability and CNS inflammation: Reviewing the connection. Nutrients 2020, 12, 3906. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.J.T.; Bagit, A.; MacPherson, R.E.K. Resveratrol, metabolic dysregulation and Alzheimer’s disease: Considerations for neurodegenerative disease. Int. J. Mol. Sci. 2021, 22, 4628. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lu, F.; Jia, X.; Yan, Q.; Zhang, X.; Mu, P. Amyloid-β (25-35) regulates neuronal damage and memory loss via SIRT1/Nrf2 in the cortex of mice. J. Chem. Neuroanat. 2021, 114, 101945. [Google Scholar] [CrossRef] [PubMed]
- Kodl, C.T.; Seaquist, E.R. Cognitive dysfunction and diabetes mellitus. Endocr. Rev. 2008, 29, 494–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahal, H.; Halama, A.; Aburima, A.; Bhagwat, A.M.; Butler, A.E.; Graumann, J.; Suhre, K.; Sathyapalan, T.; Atkin, S.L. Effect of induced hypoglycemia on inflammation and oxidative stress in type 2 diabetes and control subjects. Sci. Rep. 2020, 10, 4750. [Google Scholar] [CrossRef] [Green Version]
- Pouvreau, C.; Dayre, A.; Butkowski, E.G.; de Jong, B.; Jelinek, H.F. Inflammation and oxidative stress markers in diabetes and hypertension. J. Inflamm. Res. 2018, 11, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Da Porto, A.; Cavarape, A.; Colussi, G.; Casarsa, V.; Catena, C.; Sechi, L.A. Polyphenols rich diets and risk of type 2 diabetes. Nutrients 2021, 13, 1445. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, C.; Qiu, S.; Yuan, X.; Li, L. Effects of resveratrol on glucose control and insulin sensitivity in subjects with type 2 diabetes: Systematic review and meta-analysis. Nutr. Metab. 2017, 14, 60. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, A.; Bath, S.; Elbarbry, F. An organ system approach to explore the antioxidative, anti-Inflammatory, and cytoprotective actions of resveratrol. Oxid. Med. Cell. Longev. 2015, 2015, 803971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallauf, K.; Rimbach, G.; Rupp, P.M.; Chin, D.; Wolf, I.M. Resveratrol and lifespan in model organisms. Curr. Med. Chem. 2016, 23, 4639–4680. [Google Scholar] [CrossRef] [PubMed]
- Farhadnejad, H.; Emamat, H.; Zand, H. The effect of resveratrol on cellular senescence in normal and cancer cells: Focusing on cancer and age-related diseases. Nutr. Cancer 2019, 71, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Stromsnes, K.; Mas-Bargues, C.; Gambini, J.; Gimeno-Mallench, L. Protective effects of polyphenols present in mediterranean diet on endothelial dysfunction. Oxid. Med. Cell. Longev. 2020, 2020, 2097096. [Google Scholar] [CrossRef]
- Zordoky, B.N.; Robertson, I.M.; Dyck, J.R. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim. Biophys. Acta 2015, 1852, 1155–1177. [Google Scholar] [CrossRef] [Green Version]
- Griñán-Ferré, C.; Bellver-Sanchis, A.; Izquierdo, V.; Corpas, R.; Roig-Soriano, J.; Chillon, M.; Andres-Lacueva, C.; Somogyvari, M.; Sőti, C.; Sanfeliu, C.; et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: From antioxidant to epigenetic therapy. Ageing Res. Rev. 2021, 67, 101271. [Google Scholar] [CrossRef]
- Komorowska, J.; Wątroba, M.; Szukiewicz, D. Review of beneficial effects of resveratrol in neurodegenerative diseases such as Alzheimer’s disease. Adv. Med. Sci. 2020, 65, 415–423. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Corsetti, G. Natural compounds and autophagy: Allies against neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409. [Google Scholar] [CrossRef]
- Chimento, A.; De Amicis, F.; Sinianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to improve oral bioavailability and beneficial effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, N.A.; Guarente, L. Genetic links between diet and lifespan: Shared mechanisms from yeast to humans. Nat. Rev. Genet. 2007, 8, 835–844. [Google Scholar] [CrossRef]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [Green Version]
- Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review. Biology 2021, 10, 194. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of targeted mass spectrometry for the quantification of sirtuins in the central nervous system. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidorova-Darmos, E.; Wither, R.G.; Shulyakova, N.; Fisher, C.; Ratnam, M.; Aarts, M.; Lilge, L.; Monnier, P.P.; Eubanks, J.H. Differential expression of sirtuin family members in the developing, adult, and aged rat brain. Front Aging Neurosci. 2014, 6, 333. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Smythe, G.; Sachdev, P.; Guillermin, G.J. Differential expression of sirtuins in the aging rat brain. Front Cell. Neurosci. 2015, 9, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Luo, A.; Gao, J.; Tang, X.; Zhao, Y.; Zhou, B.; Zhou, Z.; Li, S. The role of SIRT1 in neuroinflammation and cognitive dysfunction in aged rats after anesthesia and surgery. Am. J. Transl. Res. 2019, 11, 1555–1568. [Google Scholar]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Mohan, N.; Upadhyay, A.D.; Singh, A.P.; Sahu, V.; Dwivedi, S.; Dey, A.B.; Dey, S. Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell 2014, 13, 975–980. [Google Scholar] [CrossRef]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromol. Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Han, P.; Song, M.; Nielsen, M.; Beach, T.G.; Serrano, G.E.; Liang, W.S.; Ceselli, R.J.; Shi, J. Amyloid-β increases tau by mediating sirtuin 3 in Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8592–8601. [Google Scholar] [CrossRef]
- Kumar, R.; Chaterjee, P.; Sharma, P.K.; Singh, A.K.; Gupta, A.; Gill, K.; Tripathi, M.; Dey, A.B.; Dey, S. Sirtuin1: A promising serum protein marker for early detection of Alzheimer’s disease. PLoS ONE 2013, 8, e61560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.; Mele, E.; Di Filippo, F.; Viggiano, A.; Meccariello, R. SIRT1 activity in the brain: Simultaneous effects on energy homeostasis and reproduction. Int. J. Environ. Res. Public Health 2021, 18, 1243. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef]
- Lin, S.J.; Guarente, L. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 2006, 2, e33. [Google Scholar] [CrossRef] [Green Version]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Brachmann, C.B.; Celic, I.; Kenna, M.A.; Muhammad, S.; Starai, V.J. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 2000, 97, 6658–6663. [Google Scholar] [CrossRef]
- Oberdoerffer, S.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 2010, 62, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metabol. 2009, 20, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Schug, T.T.; Li, X. Sirtuin 1 in lipid metabolism and obesity. Trends Mol. Med. 2011, 43, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Luo, J.; Zhang, H. Role of Sirtuin 1 in the pathogenesis of ocular disease. Int. J. Mol. Med. 2018, 42, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghabadi, Z.A.; Nourbakhsh, M.; Pasalar, P.; Emamgholipour, S.; Golestani, A.; Larijani, B.; Razzaghy-Azar, M. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes. Res. Clin. Pract. 2018, 12, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Kume, S.; Koya, D. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 2009, 5, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Mundt, L.A.; Shanahan, K. Graff’s Textbook of Routine Urinalysis and Body Fluids; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2010; p. 237. ISBN 978-1582558752. [Google Scholar]
- Williams-Medina, A.; Deblock, M.; Janigro, D. In vitro models of the Blood–Brain Barrier: Tools in translational medicine. Front Med. Technol. 2021, 2, 623950. [Google Scholar] [CrossRef]
- Erickson, M.A.; Wilson, M.L.; Banks, W.A. In vitro modeling of blood–brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS 2020, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Linville, R.M.; Searson, P.C. Next-generation in vitro blood–brain barrier models: Benchmarking and improving model accuracy. Fluids Barriers CNS 2021, 18, 56. [Google Scholar] [CrossRef]
- Poller, B.; Gutmann, H.; Krähenbühl, S.; Weksler, B.; Romero, I.; Cauraud, P.O.; Tuffin, G.; Drewe, J.; Huwyler, J. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 2008, 107, 1358–1368. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood-brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, N.; Yokoyama, T. Permeability of porous media: Role of the critical pore size. J. Geophys. Res. Solid Earth 2017, 122, 6955–6971. [Google Scholar] [CrossRef]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug. Des. Devel. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Yuan, Z.; Ling, H.; Fukasawa, K.; Robertson, K.; Olashaw, N.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell. Biol. 2011, 31, 4720–4734. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Lim, J.; Lee, S.; Jeong, J.; Kang, H.; Kim, Y.; Kang, J.W.; Yu, H.Y.; Jeong, E.M.; Kim, K.; et al. Sirt1 regulates DNA methylation and differentiation potential of embryonic stem cells by antagonizing Dnmt3l. Cell Rep. 2017, 18, 1930–1945. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, Q.; Wu, X.; Zhang, D.; Xing, D. Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Aβ levels in Alzheimer’s disease models. Aging Cell 2020, 19, e13054. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Émond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Min, S.-W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xiao, D.; Li, X.; Zhang, Y.; Rasouli, J.; Casella, G.; Boehm, A.; Hwang, D.; Ishikawa, L.L.; Thome, R.; et al. SIRT1 inactivation switches reactive astrocytes to an antiinflammatory phenotype in CNS autoimmunity. J. Clin. Investig. 2022, 132, e151803. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscl. Thromb. Vas. 2004, 24, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Qadirifard, M.S.; Kosari, M.; Naghsh, N.; Taei, A.A.; Klegeris, A.; Dehghani, M.; Bahrami, A.; et al. Pre-clinical studies identifying molecular pathways of neuroinflammation in Parkinson’s disease: A systematic review. Front. Aging Neurosci. 2022, 14, 855776. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.R.; Pogue, A.I.; Sharfman, N.M.; Taylor, C.; Lukiw, W.J. Lipopolysaccharides (LPSs) as potent neurotoxic glycolipids in Alzheimer’s disease (AD). Int. J. Mol. Sci. 2022, 23, 12671. [Google Scholar] [CrossRef]
- Takashiba, S.; Van Dyke, T.E.; Amar, S.; Murayama, Y.; Soskolne, A.W.; Shapira, L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kB. Infect. Immun. 1999, 67, 5573–5578. [Google Scholar] [CrossRef] [Green Version]
- Berman, R.S.; Frew, J.D.; Martin, W. Endotoxin-induced arterial endothelial barrier dysfunction assessed by an in vitro model. Br. J. Pharmacol. 1993, 110, 1282–1284. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.; Fernandis, A.Z.; Cherla, R.P.; Park, I.W.; Ganju, R.K. Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J. Immunol. 2002, 168, 5860–5866. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.L.; Lee, S.C. Distinct patterns of stimulus-inducible chemokine mRNA accumulation in human fetal astrocytes and microglia. Glia 2000, 30, 74–81. [Google Scholar] [CrossRef]
- Solomon, K.R.; Kurt-Jones, E.A.; Saladino, R.A.; Stack, A.M.; Dunn, I.F.; Ferretti, M.; Golenbock, D.; Fleisher, G.R.; Finberg, R.W. Heterotrimeric G proteins physically associated with the lipopolysaccharide receptor CD14 modulate both in vivo and in vitro responses to lipopolysaccharide. J. Clin. Investig. 1998, 102, 2019–2027. [Google Scholar] [CrossRef] [Green Version]
- Borzęcka, K.; Płóciennikowska, A.; Björkelund, H.; Sobota, A.; Kwiatkowska, K. CD14 mediates binding of high doses of LPS but is dispensable for TNF-α production. Mediat. Inflamm. 2013, 2013, 824919. [Google Scholar] [CrossRef] [Green Version]
- Ngkelo, A.; Meja, K.; Yeadon, M.; Adcock, I.; Kirkham, P.A. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3 kinase signalling. J. Inflamm. 2012, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Chen, G.; Qi, M.; El-Assaad, F.; Wang, Y.; Dong, S.; Chen, L.; Yu, D.; Weaver, J.C.; Beretov, J.; et al. Gram negative bacterial inflammation ameliorated by the Plasma Protein Beta 2-Glycoprotein I. Sci. Rep. 2016, 6, 33656. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.J.; Hume, D.A. Endotoxin signal transduction in macrophages. J. Leukoc. Biol. 1996, 60, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.V. The effect of bacterial pyrogen on the blood-brain barrier permeability for trypan blue. J. Pathol. Bacteriol. 1965, 89, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Eckman, P.L.; King, W.M.; Brunson, J.G. Studies on the blood brain barrier. I. Effects produced by a single injection of Gram-negative endotoxin on the permeability of the cerebral vessels. Am. J. Pathol. 1958, 34, 631–643. [Google Scholar]
- Wispelwey, B.; Lesse, A.J.; Hansen, E.J.; Scheld, W.M. Haemophilus influenzae lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J. Clin. Investig. 1988, 82, 1339–1346. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. LPS-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neuromuscular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S.; Lachance, C.; Patrizi, S.; Lefebvre, S.; Follett, P.L.; Jensen, F.E.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 2002, 22, 2478–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauwels, A.; Frei, K.; Sansano, S.; Fearns, C.; Ulevitch, R.; Zimmerli, W.; Landmann, R. The origin and function of soluble CD14 in experimental bacterial meningitis. J. Immunol. 1999, 162, 4762–4772. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, K.H.; Park, H.S.; Choi, S.J. LPS sensing mechanism of human astrocytes: Evidence of functional TLR4 expression and requirement of soluble CD14. J. Bacteriol. Virol. 2017, 47, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, Y.; Guo, X.; Zeng, Z.; Wu, J.; Liu, Y.; He, J.; Wang, R.; Huang, Q.; Chen, Z. Sirt1 protects endothelial cells against LPS-induced barrier dysfunction. Oxid. Med. Cell. Longev. 2017, 2017, 4082102. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.A.; Yao, F.; Wong, J.J.; George, J.; Xu, H.; Chiu, K.P.; Sung, W.K.; Lipovich, L.; Vega, V.B.; Chen, J.; et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell 2007, 27, 622–635. [Google Scholar] [CrossRef]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [Green Version]
- Young, H.A. Regulation of interferon-γ gene expression. J. Interf. Cytokine Res. 1996, 16, 563–568. [Google Scholar] [CrossRef]
- Sica, A.; Dorman, L.; Viggiano, V.; Cippitelli, M.; Ghosh, P.; Rice, N.; Young, H.A. Interaction of NF-κ and NFAT with the interferon-γ promoter. J. Biol. Chem. 1997, 272, 30412–30420. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhao, Y.; Wu, X.; Xia, M.; Fang, M.; Iwasaki, Y.; Sha, J.; Chen, Q.; Xu, Y.; Shen, A. Interferon gamma (IFN-γ) disrupts energy expenditure and metabolic homeostasis by suppressing SIRT1 transcription. Nucl. Acids Res. 2012, 40, 1609–1620. [Google Scholar] [CrossRef]
- Yamakushi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22phox by NF-κB in human aortic smooth muscle cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an anti-inflammatory and anti-aging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Resp Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, L. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lowry, S.F.; Guarente, L.; Haimovich, B. Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J. Biol. Chem. 2010, 285, 41391–41401. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation end-products (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GADPH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between oxidative stress and SIRT1: Impact on the aging process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κ and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.A.; Carcillo, J.A.; Szabo, C.; Clark, R.S. Intra-mitochondrial Poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, A.; Tada-Oikawa, S.; Kawanishi, S.; Oikawa, S. H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell. Physiol. Biochem. 2007, 20, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) Polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Wada-Hiraike, O.; Yano, T.; Shirane, A.; Hirano, M.; Hiraike, H.; Koyama, S.; Oishi, H.; Yoshino, O.; Miyamoto, Y.; et al. Resveratrol promotes expression of SIRT1 and StAR in rat ovarian granulosa cells: An implicative role of SIRT1 in the ovary. Reprod. Biol. Endocrinol. 2012, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.P.; Singh, N.P.; Singh, B.; Hofseth, L.J.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Resveratrol (trans-3,5,4′-trihydroxystilbene) induces silent mating type information regulation-1 and down-regulates nuclear transcription factor-kappaB activation to abrogate dextran sulfate sodium-induced colitis. J. Pharmacol. Exp. Ther. 2010, 332, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [Green Version]
- Suchankova, G.; Nelson, L.E.; Gerhart-Hines, Z.; Kelly, M.; Gauthier, M.S.; Saha, A.K.; Ido, Y.; Puigserver, P.; Ruderman, N.B. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem. Biophys. Res. Commun. 2009, 378, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Um, J.H.; Park, S.J.; Kang, H.; Yang, S.; Foretz, M.; McBurney, M.W.; Kim, M.K.; Viollet, B.; Chung, J.H. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010, 59, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Chen, X.; Song, X.; Chen, Y.; Jia, R.; Zou, Y.; Li, L.; Yin, L.; He, C.; Liang, X.; et al. Resveratrol inhibits LPSinduced inflammation through suppressing the signaling cascades of TLR4NFκB/MAPKs/IRF3. Exp. Ther. Med. 2020, 19, 1824–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ma, Z.; Zou, W.; Guo, H.; Liu, M.; Ma, Y.; Zhang, L. The appropriate marker for astrocytes: Comparing the distribution and expression of three astrocytic markers in different mouse cerebral regions. BioMed. Res Int. 2019, 2019, 9605265. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.C.; Morrison, E.H. Histology, Astrocytes. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Newman, E.A. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos. Trans. R. Soc. Lond B Biol. Sci. 2015, 370, 20140195. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2016, 18, 31–41. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, X.; Liu, T.; Liu, H.; Tong, L.; Jia, S.; Wang, Y.F. Neurochemical regulation of the expression and function of glial fibrillary acidic protein in astrocytes. Glia 2020, 68, 878–897. [Google Scholar] [CrossRef]
- Amalia, L. Glial Fibrillary Acidic Protein (GFAP): Neuroinflammation biomarker in acute ischemic stroke. J. Inflamm. Res. 2021, 14, 7501–7506. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [Green Version]
- Ikeshima-Kataoka, H. Neuroimmunological implications of AQP4 in astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.C.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A.; Norenberg, M.D. Primary cultures of astrocytes: Their value in understanding astrocytes in health and disease. Neurochem. Res. 2012, 37, 2569–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Romero, M.C.; Argüelles, S.; Villarán, R.F.; de Pablos, R.M.; Delgado-Cortés, M.J.; Santiago, M.; Herrera, A.J.; Cano, J.; Machado, A. Simvastatin prevents the inflammatory process and the dopaminergic degeneration induced by the intranigral injection of lipopolysaccharide. J. Neurochem. 2008, 105, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Deng, I.; Corrigan, F.; Zhai, G.; Zhou, X.F.; Bobrovskaya, L. Lipopolysaccharide animal models of Parkinson’s disease: Recent progress and relevance to clinical disease. Brain. Behav. Immun. Health 2020, 4, 100060. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Abraham, J.; Johnson, R.W. Consuming a diet supplemented with resveratrol reduced infection-related neuroinflammation and deficits in working memory in aged mice. Rejuvenation Res. 2009, 12, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Terrando, N.; Rei Fidalgo, A.; Vizcaychipi, M.; Cibelli, M.; Ma, D.; Monaco, C.; Feldmann, M.; Maze, M. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit. Care 2010, 14, R88. [Google Scholar] [CrossRef] [Green Version]
- Lasselin, J.; Schedlowski, M.; Karshikoff, B.; Engler, H.; Lekander, M.; Konsman, J.P. Comparison of bacterial lipopolysaccharide-induced sickness behavior in rodents and humans: Relevance for symptoms of anxiety and depression. Neurosci. Biobehav. Rev. 2020, 115, 15–24. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Du, X.M.; Ma, X.J.; Zong, Y.; Chen, J.K.; Yu, C.L.; Liu, Y.G.; Chen, Y.C.; Zhao, L.J.; Lu, G.C. Activation of the NLRP3 inflammasome in lipopolysaccharide-induced mouse fatigue and its relevance to chronic fatigue syndrome. J. Neuroinflammation 2016, 13, 71. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.L.; Shi, D.L.; Gu, H.Y.; Zheng, M.Z.; Hu, J.; Song, X.H.; Shen, Y.L.; Chen, Y.Y. Resveratrol attenuates inflammatory hyperalgesia by inhibiting glial activation in mice spinal cords. Mol. Med. Rep. 2016, 13, 4051–4057. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.H.; Wang, L.L. Diffusion efficiency and bioavailability of resveratrol administered to rat brain by different routes: Therapeutic implications. Neurotherapeutics 2015, 12, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Shu, X.H.; Wu, M.L.; Zheng, X.; Jia, B.; Kong, Q.Y.; Liu, J.; Li, H. Postoperative resveratrol administration improves prognosis of rat orthotopic glioblastomas. BMC Cancer 2018, 18, 871. [Google Scholar] [CrossRef]
- Gambini, J.; Ingles, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of resveratrol: In vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug. Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.; Tuor, U.I.; Thompson, R.; Institoris, A.; Kulynych, A.; Zhang, X.; Kinniburgh, D.W.; Bari, F.; Busija, D.W.; Barber, P.A. Protection against recurrent stroke with resveratrol: Endothelial protection. PLoS ONE 2012, 7, e47792. [Google Scholar] [CrossRef] [Green Version]
- Song, F.H.; Liu, D.Q.; Zhou, Y.Q.; Mei, W. SIRT1: A promising therapeutic target for chronic pain. CNS Neurosci. Ther. 2022, 28, 818–828. [Google Scholar] [CrossRef]
- Joseph, A.; Balakrishnan, A.; Shanmughan, P.; Maliakel, B.; Madhavamenon, K.I. Micelle/hydrogel composite as a “Natural Self-Emulsifying Reversible Hybrid Hydrogel (N’SERH)” enhances the oral bioavailability of free (unconjugated) resveratrol. ACS Omega 2022, 7, 12835–12845. [Google Scholar] [CrossRef]
- Almeida, T.C.; Seibert, J.B.; de Souza Almeida, S.H.; Amparo, T.R.; de Medeiros Teixeira, L.F.; Barichello, J.M.; Postacchini, B.B.; dos Santos, O.D.H.; da Silva, G.N. Polymeric micelles containing resveratrol: Development, characterization, cytotoxicity on tumor cells and antimicrobial activity. Braz. J. Pharm Sci. 2020, 56, e18411. [Google Scholar] [CrossRef]
- Chimento, A.; Santarsiero, A.; Iacopetta, D.; Ceramella, J.; De Luca, A.; Infantino, V.; Parisi, O.I.; Avena, P.; Bonomo, M.G.; Saturnino, C.; et al. A Phenylacetamide resveratrol derivative exerts inhibitory effects on breast cancer cell growth. Int. J. Mol. Sci. 2021, 22, 5255. [Google Scholar] [CrossRef]
- Tu, C.; Fiandalo, M.V.; Pop, E.; Stocking, J.J.; Azabdaftari, G.; Li, J.; Wei, H.; Ma, D.; Qu, J.; Mohler, J.L.; et al. Proteomic analysis of charcoal-stripped Fetal Bovine Serum reveals changes in the Insulin-like Growth Factor signaling pathway. J. Proteome Res. 2018, 17, 2963–2977. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Voisin, P.; Bouchaud, V.; Bezancon, E.; Franconi, J.M.; Pellerin, L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: A comparative NMR study. Eur. J. Neurosci. 2006, 24, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Komorowska, J.; Wątroba, M.; Bednarzak, M.; Grabowska, A.D.; Szukiewicz, D. The role of glucose concentration and resveratrol in modulating neuroinflammatory cytokines: Insights from an in vitro blood-brain barrier model. Med. Sci. Mon. 2023; paper in press. [Google Scholar]
- Wight, R.D.; Tull, C.A.; Deel, M.W.; Stroope, B.L.; Eubanks, A.G.; Chavis, J.A.; Drew, P.D.; Hensley, L.L. Resveratrol effects on astrocyte function: Relevance to neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2012, 426, 112–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc. 2008, 2008, pdb.prot4986. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowska, A.D.; Wątroba, M.; Witkowska, J.; Mikulska, A.; Sepúlveda, N.; Szukiewicz, D. Interplay between Systemic Glycemia and Neuroprotective Activity of Resveratrol in Modulating Astrocyte SIRT1 Response to Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 11640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241411640
Grabowska AD, Wątroba M, Witkowska J, Mikulska A, Sepúlveda N, Szukiewicz D. Interplay between Systemic Glycemia and Neuroprotective Activity of Resveratrol in Modulating Astrocyte SIRT1 Response to Neuroinflammation. International Journal of Molecular Sciences. 2023; 24(14):11640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241411640
Chicago/Turabian StyleGrabowska, Anna D., Mateusz Wątroba, Joanna Witkowska, Agnieszka Mikulska, Nuno Sepúlveda, and Dariusz Szukiewicz. 2023. "Interplay between Systemic Glycemia and Neuroprotective Activity of Resveratrol in Modulating Astrocyte SIRT1 Response to Neuroinflammation" International Journal of Molecular Sciences 24, no. 14: 11640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241411640