
Partial Loss of Function ABCA12 Mutations Generate Reduced Deposition of Glucosyl-Ceramide, Leading to Patchy Ichthyosis and Erythrodermia Resembling Erythrokeratodermia Variabilis et Progressiva (EKVP)
 , , , ,
, , , ,  and
and 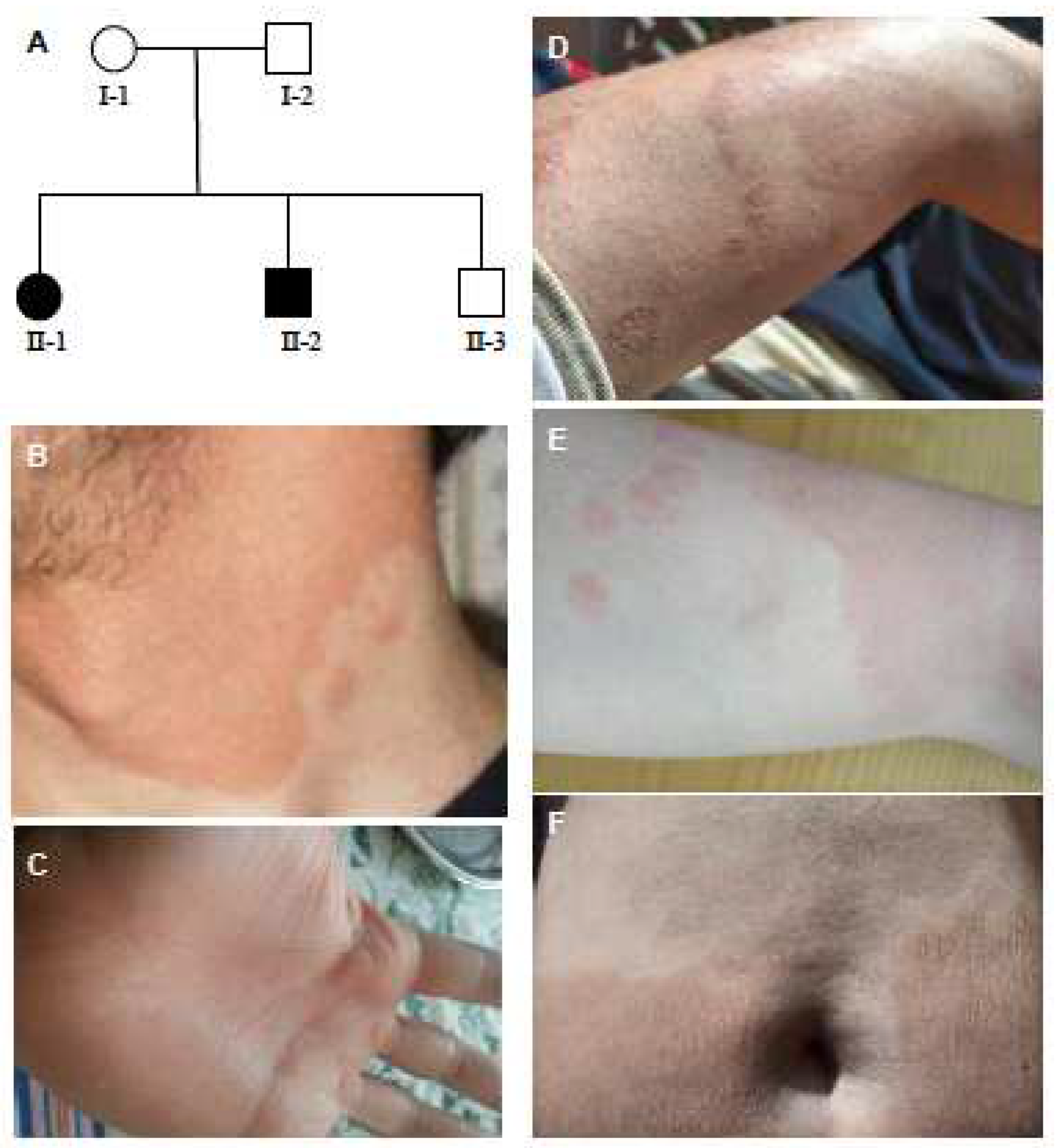{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Patient Presentation
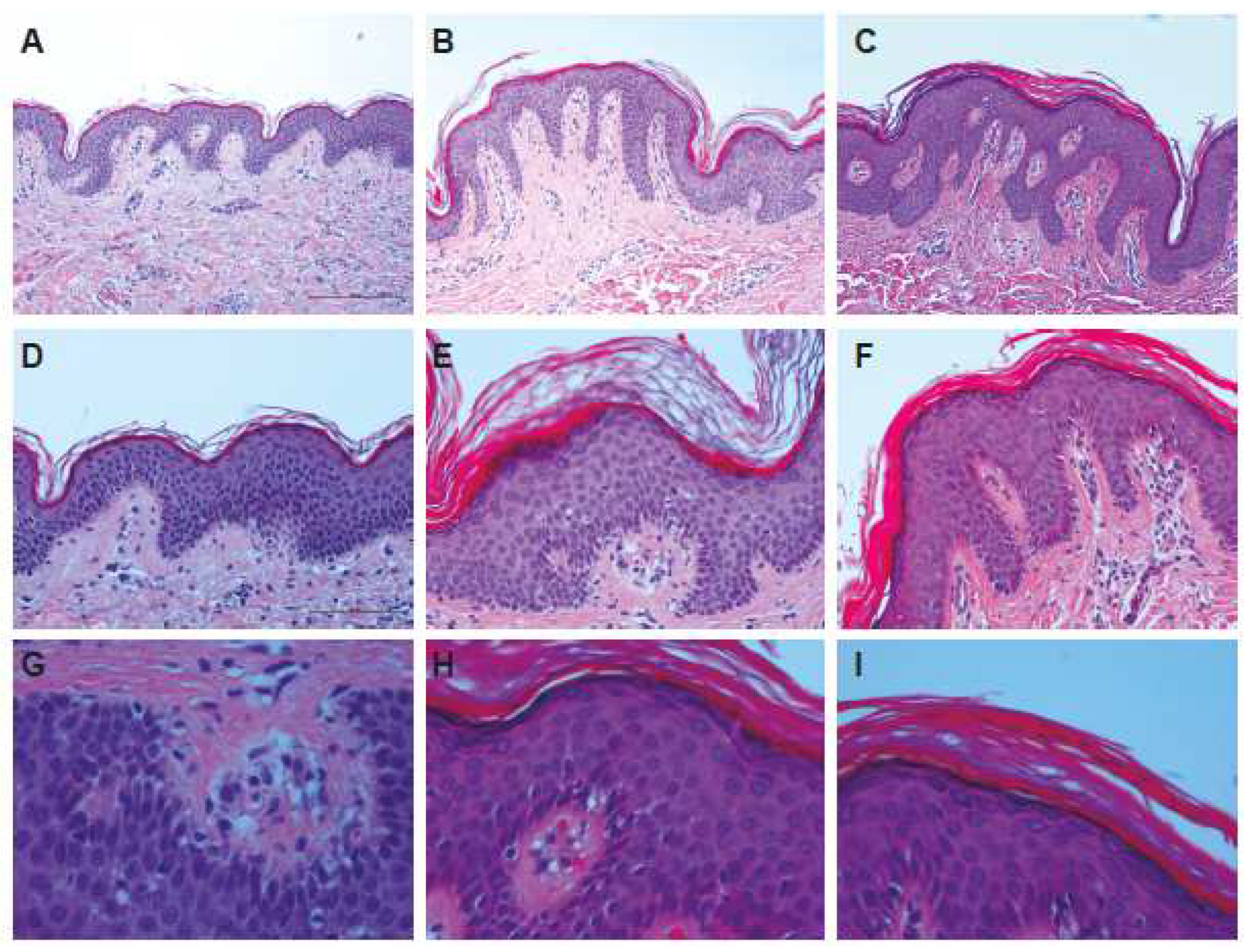
2.2. Histological Examination
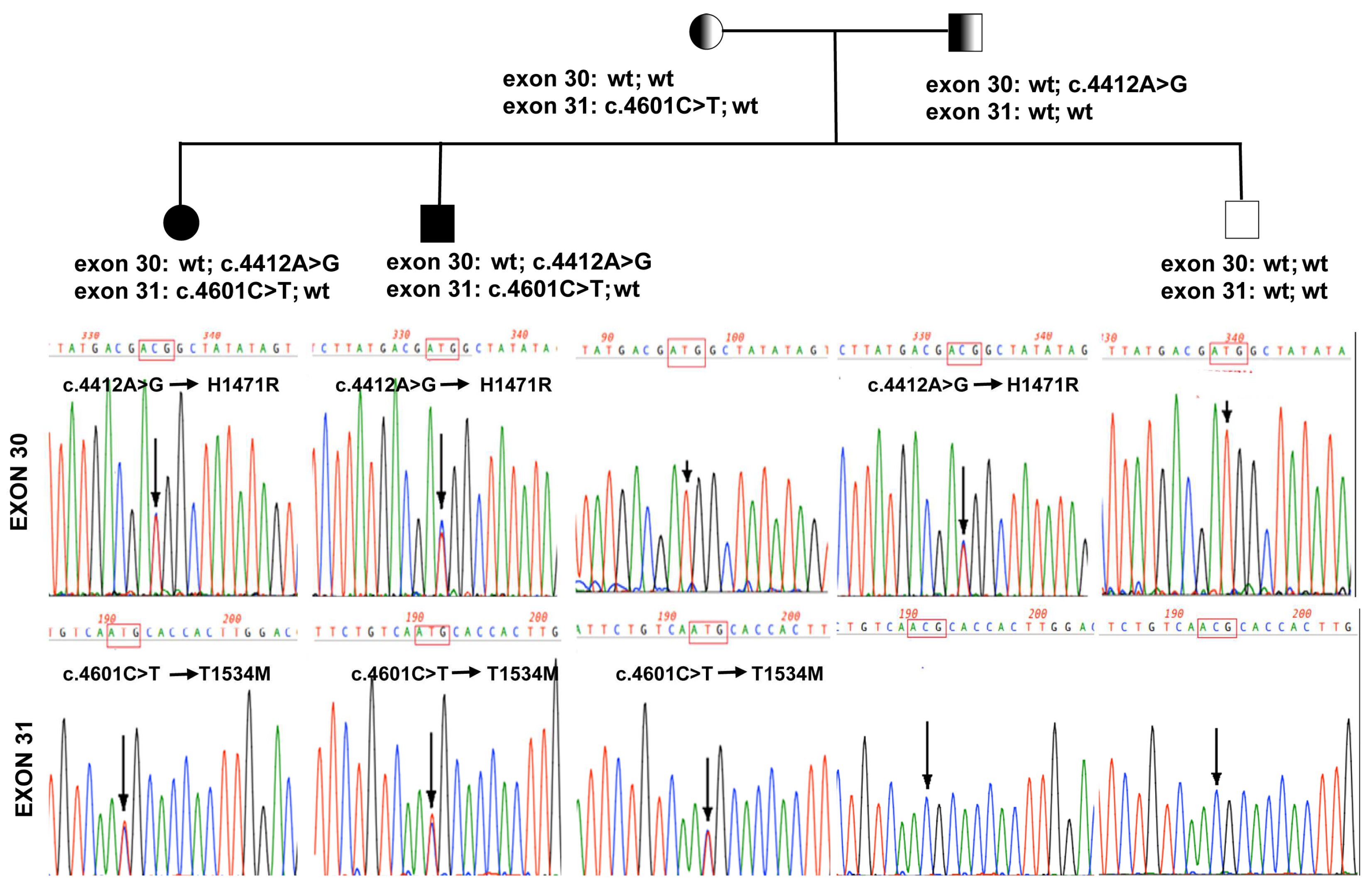
2.3. Genetic Analysis
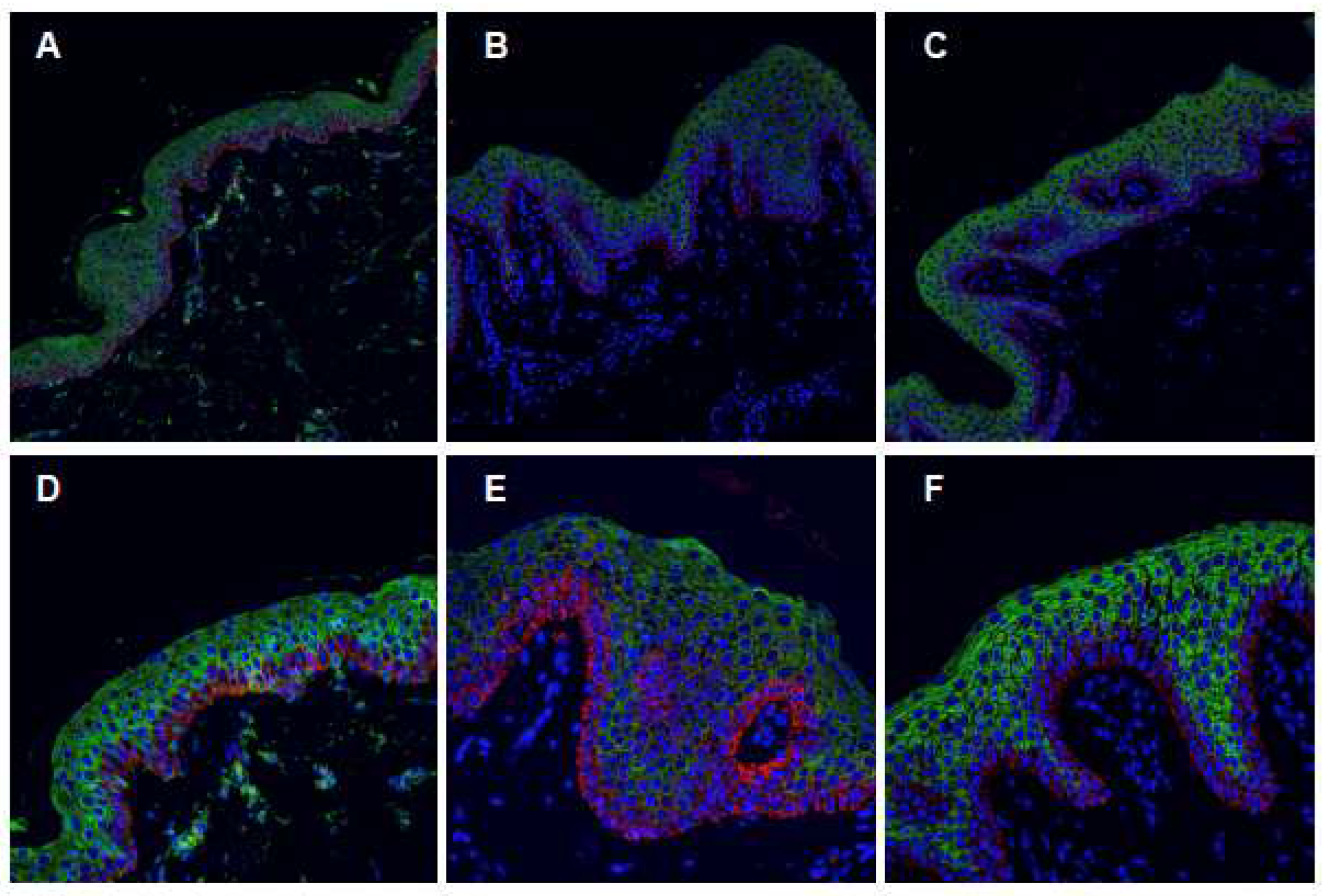
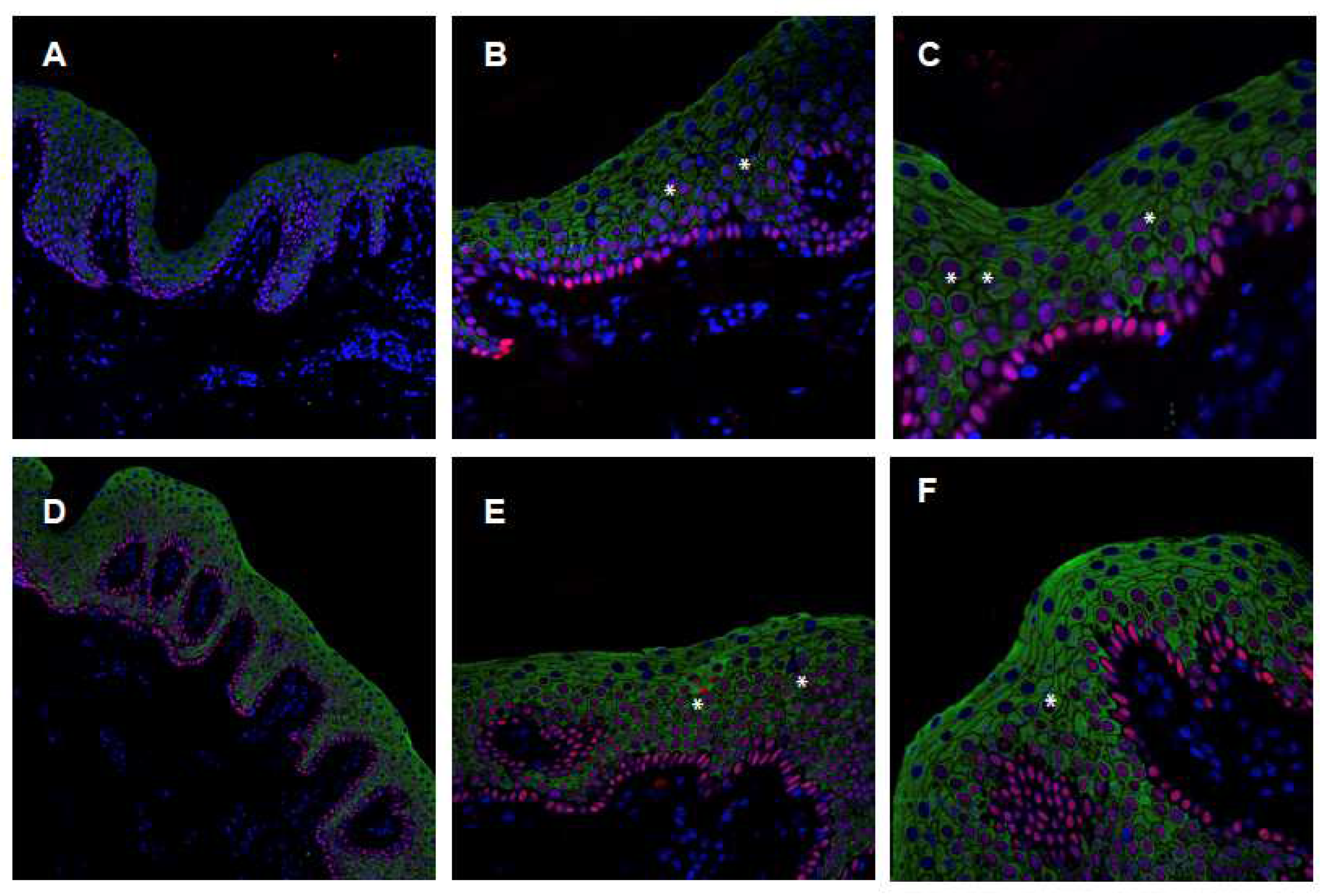
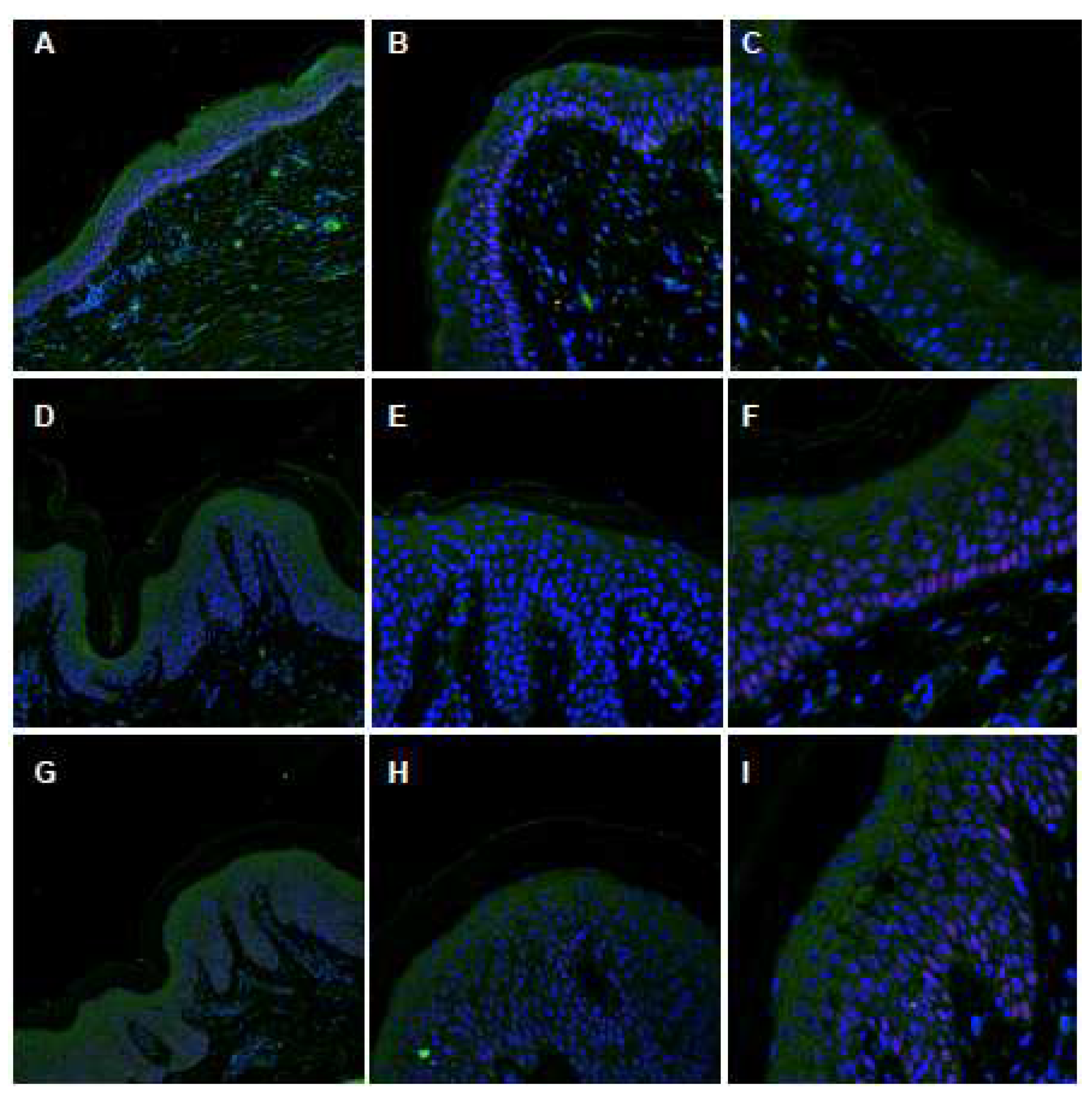
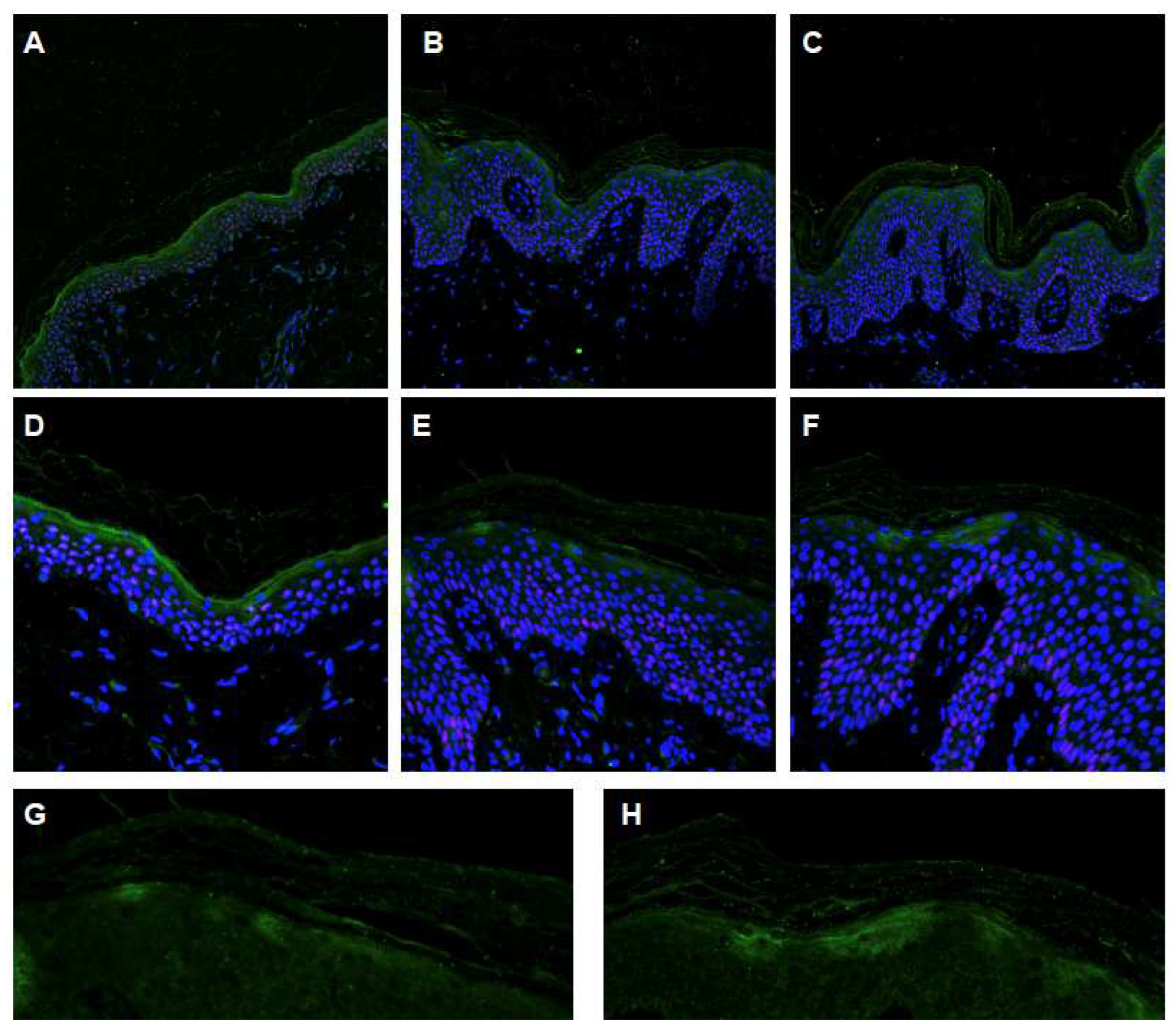
2.4. Characterization of the ABCA12 Physiological Modifications
3. Discussion
4. Materials and Methods
4.1. Genetic Analysis
Exome Sequencing
4.2. Light Microscopy
4.3. Confocal Immunofluorescence Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fischer, J.; Bourrat, E. Genetics of Inherited Ichthyoses and Related Diseases. Acta Derm. -Venereol. 2020, 100, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Youssefian, L.; Saeidian, A.H.; Vahidnezhad, H. Molecular Genetics of Keratinization Disorders—What’s New About Ichthyosis. Acta Derm. -Venereol. 2020, 100, adv00095. [Google Scholar] [CrossRef] [PubMed]
- Arin, M.J.; Oji, V.; Emmert, S.; Hausser, I.; Traupe, H.; Krieg, T.; Grimberg, G. Expanding the keratin mutation database: Novel and recurrent mutations and genotype-phenotype correlations in 28 patients with epidermolytic ichthyosis. Br. J. Dermatol. 2011, 164, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Corden, L.D.; McLean, W.H. Human keratin diseases: Hereditary fragility of specific epithelial tissues. Exp. Dermatol. 1996, 5, 297–307. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H. Human keratin diseases: The increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br. J. Dermatol. 1999, 140, 815–828. [Google Scholar] [CrossRef]
- Lane, E.B.; McLean, W.H. Keratins and skin disorders. J. Pathol. 2004, 204, 355–366. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef]
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Soreze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef]
- Goldsmith, L.A. The ichthyoses. Prog. Med. Genet. 1976, 1, 185–210. [Google Scholar]
- Akiyama, M.; Sugiyama-Nakagiri, Y.; Sakai, K.; McMillan, J.R.; Goto, M.; Arita, K.; Tsuji-Abe, Y.; Tabata, N.; Matsuoka, K.; Sasaki, R.; et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J. Clin. Investig. 2005, 115, 1777–1784. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Norgett, E.E.; Unsworth, H.; Teh, M.T.; Cullup, T.; Mein, C.A.; Dopping-Hepenstal, P.J.; Dale, B.A.; Tadini, G.; Fleckman, P.; et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am. J. Hum. Genet. 2005, 76, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Van Leersum, F.S.; Seyger, M.M.B.; Theunissen, T.E.J.; Bongers, E.; Steijlen, P.M.; van Geel, M. Recessive mosaicism in ABCA12 causes blaschkoid congenital ichthyosiform erythroderma. Br. J. Dermatol. 2020, 182, 208–211. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; McGrath, J.A.; Lam, H.; Iizuka, H.; Friedman, R.A.; Christiano, A.M. The molecular pathology of progressive symmetric erythrokeratoderma: A frameshift mutation in the loricrin gene and perturbations in the cornified cell envelope. Am. J. Hum. Genet. 1997, 61, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Smith, L.E.; Bailey, R.A.; Itin, P.; Hohl, D.; Epstein, E.H., Jr.; DiGiovanna, J.J.; Compton, J.G.; Bale, S.J. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat. Genet. 1998, 20, 366–369. [Google Scholar] [CrossRef]
- Macari, F.; Landau, M.; Cousin, P.; Mevorah, B.; Brenner, S.; Panizzon, R.; Schorderet, D.F.; Hohl, D.; Huber, M. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am. J. Hum. Genet. 2000, 67, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Brown, N.; Rouan, F.; Van der Schroeff, J.G.; Bijlsma, E.; Eichenfield, L.F.; Sybert, V.P.; Greer, K.E.; Hogan, P.; Campanelli, C.; et al. Genetic heterogeneity in erythrokeratodermia variabilis: Novel mutations in the connexin gene GJB4 (Cx30.3) and genotype-phenotype correlations. J. Investig. Dermatol. 2003, 120, 601–609. [Google Scholar] [CrossRef]
- Boyden, L.M.; Craiglow, B.G.; Zhou, J.; Hu, R.; Loring, E.C.; Morel, K.D.; Lauren, C.T.; Lifton, R.P.; Bilguvar, K.; Yale Center for Mendelian Genomics; et al. Dominant De Novo Mutations in GJA1 Cause Erythrokeratodermia Variabilis et Progressiva, without Features of Oculodentodigital Dysplasia. J. Investig. Dermatol. 2015, 135, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Leta, A.; Pedicelli, C.; Candi, E.; Ranalli, M.; Puddu, P.; Paradis, M.; Angelo, C.; Bagetta, G.; Melino, G. A novel recessive connexin 31 (GJB3) mutation in a case of erythrokeratodermia variabilis. J. Investig. Dermatol. 2004, 122, 837–839. [Google Scholar] [CrossRef]
- Fuchs-Telem, D.; Pessach, Y.; Mevorah, B.; Shirazi, I.; Sarig, O.; Sprecher, E. Erythrokeratoderma variabilis caused by a recessive mutation in GJB3. Clin. Exp. Dermatol. 2011, 36, 406–411. [Google Scholar] [CrossRef]
- Terrinoni, A.; Melino, G. Recessive EKV. J. Investig. Dermatol. 2005, 124, 270–271. [Google Scholar] [CrossRef]
- Gottfried, I.; Landau, M.; Glaser, F.; Di, W.L.; Ophir, J.; Mevorah, B.; Ben-Tal, N.; Kelsell, D.P.; Avraham, K.B. A mutation in GJB3 is associated with recessive erythrokeratodermia variabilis (EKV) and leads to defective trafficking of the connexin 31 protein. Hum. Mol. Genet. 2002, 11, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential roles of p63 isoforms in epidermal development: Selective genetic complementation in p63 null mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Palombo, R.; Pitolli, C.; Caporali, S.; De Berardinis, R.; Ciccarone, S.; Lanzillotta, A.; Mauramati, S.; Porta, G.; Minieri, M.; et al. Role of the TAp63 Isoform in Recurrent Nasal Polyps. Folia Biol. 2019, 65, 170–180. [Google Scholar]
- Serra, V.; Castori, M.; Paradisi, M.; Bui, L.; Melino, G.; Terrinoni, A. Functional characterization of a novel TP63 mutation in a family with overlapping features of Rapp-Hodgkin/AEC/ADULT syndromes. Am. J. Med. Genet. Part A 2011, 155, 3104–3109. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P.; Francis, J.S.; Corden, L.D.; Smith, L.T.; Weaver, M.; Stephens, K.; McLean, W.H. Cyclic ichthyosis with epidermolytic hyperkeratosis: A phenotype conferred by mutations in the 2B domain of keratin K1. Am. J. Hum. Genet. 1999, 64, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Joh, G.Y.; Traupe, H.; Metze, D.; Nashan, D.; Huber, M.; Hohl, D.; Longley, M.A.; Rothnagel, J.A.; Roop, D.R. A novel dinucleotide mutation in keratin 10 in the annular epidermolytic ichthyosis variant of bullous congenital ichthyosiform erythroderma. J. Investig. Dermatol. 1997, 108, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Cullup, T.; Norgett, E.E.; Hill, T.; Barton, S.; Dale, B.A.; Sprecher, E.; Sheridan, E.; Taylor, A.E.; Wilroy, R.S.; et al. ABCA12 is the major harlequin ichthyosis gene. J. Investig. Dermatol. 2006, 126, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim. Et Biophys. Acta 2014, 1841, 435–440. [Google Scholar] [CrossRef]
- Mitsutake, S.; Suzuki, C.; Akiyama, M.; Tsuji, K.; Yanagi, T.; Shimizu, H.; Igarashi, Y. ABCA12 dysfunction causes a disorder in glucosylceramide accumulation during keratinocyte differentiation. J. Dermatol. Sci. 2010, 60, 128–129. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, S.H. Epidermal permeability barrier defects and barrier repair therapy in atopic dermatitis. Allergy Asthma Immunol. Res. 2014, 6, 276–287. [Google Scholar] [CrossRef]
- Albanesi, C.; Scarponi, C.; Giustizieri, M.L.; Girolomoni, G. Keratinocytes in inflammatory skin diseases. Curr. Drug Targets-Inflamm. Allergy 2005, 4, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y. Molecular Mechanism of Epidermal Barrier Dysfunction as Primary Abnormalities. Int. J. Mol. Sci. 2020, 21, 1194. [Google Scholar] [CrossRef] [PubMed]
- Codispoti, A.; Colombo, E.; Zocchi, L.; Serra, V.; Pertusi, G.; Leigheb, G.; Tiberio, R.; Bornacina, G.; Zuccoli, R.; Ramponi, A.; et al. Knuckle pads, in an epidermal palmoplantar keratoderma patient with Keratin 9 R163W transgrediens expression. Eur. J. Dermatol. 2009, 19, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Puddu, P.; Didona, B.; De Laurenzi, V.; Candi, E.; Smith, F.J.D.; McLean, W.H.I.; Melino, G. A Mutation in the V1 Domain of K16 is Responsible for Unilateral Palmoplantar Verrucous Nevus. J. Investig. Dermatol. 2000, 114, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Smith, F.J.; Didona, B.; Canzona, F.; Paradisi, M.; Huber, M.; Hohl, D.; David, A.; Verloes, A.; Leigh, I.M.; et al. Novel and recurrent mutations in the genes encoding keratins K6a, K16 and K17 in 13 cases of pachyonychia congenita. J. Investig. Dermatol. 2001, 117, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Giardina, E.; Pertusi, G.; Cascella, R.; Serra, V.; Bornacina, C.; Palombo, R.; Tiberio, R.; Gattoni, M.; Novelli, G.; et al. Absence of filaggrin mutation in a patient affected by pachyonychia congenita and mild atopic dermatitis. Eur. J. Dermatol. 2014, 24, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Didona, B.; Caporali, S.; Chillemi, G.; Lo Surdo, A.; Paradisi, M.; Annichiarico-Petruzzelli, M.; Candi, E.; Bernardini, S.; Melino, G. Role of the keratin 1 and keratin 10 tails in the pathogenesis of ichthyosis hystrix of Curth Macklin. PLoS ONE 2018, 13, e0195792. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrinoni, A.; Sala, G.; Bruno, E.; Pitolli, C.; Minieri, M.; Pieri, M.; Gambacurta, A.; Campione, E.; Belardi, R.; Bernardini, S. Partial Loss of Function ABCA12 Mutations Generate Reduced Deposition of Glucosyl-Ceramide, Leading to Patchy Ichthyosis and Erythrodermia Resembling Erythrokeratodermia Variabilis et Progressiva (EKVP). Int. J. Mol. Sci. 2023, 24, 13962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241813962
Terrinoni A, Sala G, Bruno E, Pitolli C, Minieri M, Pieri M, Gambacurta A, Campione E, Belardi R, Bernardini S. Partial Loss of Function ABCA12 Mutations Generate Reduced Deposition of Glucosyl-Ceramide, Leading to Patchy Ichthyosis and Erythrodermia Resembling Erythrokeratodermia Variabilis et Progressiva (EKVP). International Journal of Molecular Sciences. 2023; 24(18):13962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241813962
Chicago/Turabian StyleTerrinoni, Alessandro, Gabriele Sala, Ernesto Bruno, Consuelo Pitolli, Marilena Minieri, Massimo Pieri, Alessandra Gambacurta, Elena Campione, Riccardo Belardi, and Sergio Bernardini. 2023. "Partial Loss of Function ABCA12 Mutations Generate Reduced Deposition of Glucosyl-Ceramide, Leading to Patchy Ichthyosis and Erythrodermia Resembling Erythrokeratodermia Variabilis et Progressiva (EKVP)" International Journal of Molecular Sciences 24, no. 18: 13962. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241813962