
Ketogenic Diet Affects Sleep Architecture in C57BL/6J Wild Type and Fragile X Mice

Abstract
:1. Introduction
2. Results
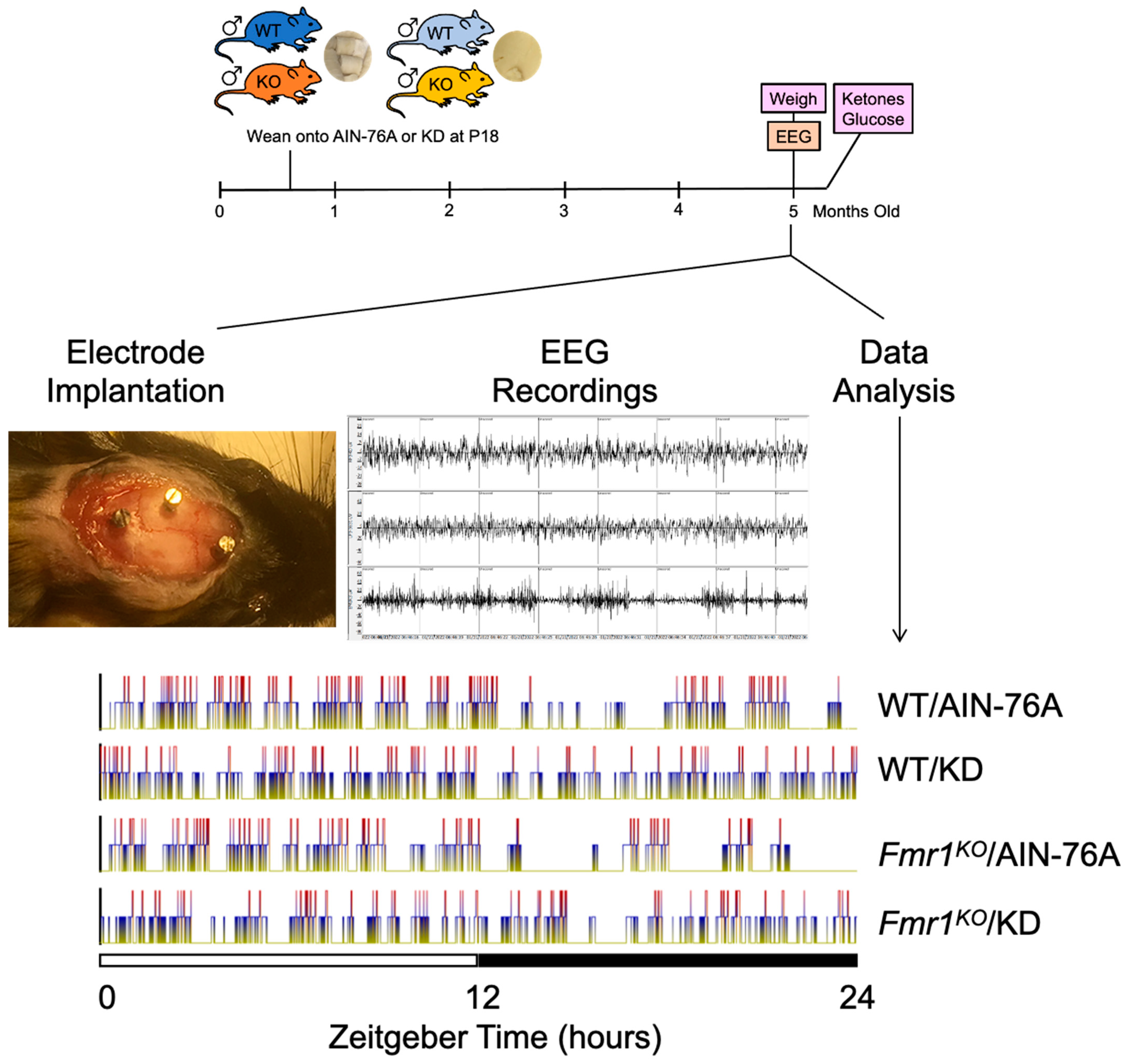
2.1. Study Design
2.2. Differences in 24-h Vigilance State Distribution Are Diet Dependent
2.3. KD Increases Time Spent in Sleep during the Dark Cycle
2.4. Sleep EEG Selectively Correlates with Actigraphy Findings during the Dark Cycle
2.5. KD Increases the Number of Wake, NREM and REM Bouts in WT Mice during the Dark Cycle
2.6. KD Decreases the Length of Wake Bouts in Mice during the Dark Cycle
2.7. Fmr1KO Mice Do Not Exhibit Epileptic Activity on Sleep EEG
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mice
4.3. EEG Electrode Implantation
4.4. Biometrics
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: A model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 1994, 78, 23–33. [Google Scholar]
- Berry-Kravis, E.; Hessl, D.; Abbeduto, L.; Reiss, A.L.; Beckel-Mitchener, A.; Urv, T.K.; Groups, O.M.W. Outcome measures for clinical trials in fragile X syndrome. J. Dev. Behav. Pediatr. 2013, 34, 508–522. [Google Scholar] [CrossRef]
- Westmark, P.R.; Gutierrez, A.; Gholston, A.K.; Wilmer, T.M.; Westmark, C.J. Preclinical testing of the ketogenic diet in fragile X mice. Neurochem. Int. 2020, 134, 104687. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139 (Suppl. S3), S194–S206. [Google Scholar] [CrossRef]
- Angriman, M.; Caravale, B.; Novelli, L.; Ferri, R.; Bruni, O. Sleep in children with neurodevelopmental disabilities. Neuropediatrics 2015, 46, 199–210. [Google Scholar] [CrossRef]
- Kronk, R.; Bishop, E.E.; Raspa, M.; Bickel, J.O.; Mandel, D.A.; Bailey, D.B. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 2010, 33, 679–687. [Google Scholar] [CrossRef]
- Kronk, R.; Dahl, R.; Noll, R. Caregiver reports of sleep problems on a convenience sample of children with fragile X syndrome. Am. J. Intellect. Dev. Disabil. 2009, 114, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.L.; Loesch, D.Z.; Martin, M.J.; Hagerman, R.J.; Armstrong, S.M.; Huggins, R.M. Melatonin profiles and sleep characteristics in boys with fragile X syndrome: A preliminary study. Am. J. Med. Genet. 2000, 95, 307–315. [Google Scholar] [CrossRef]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef]
- Agar, G.; Brown, C.; Sutherland, D.; Coulborn, S.; Oliver, C.; Richards, C. Sleep disorders in rare genetic syndromes: A meta-analysis of prevalence and profile. Mol. Autism 2021, 12, 18. [Google Scholar] [CrossRef]
- Budimirovic, D.B.; Protic, D.D.; Delahunty, C.M.; Andrews, H.F.; Choo, T.H.; Xu, Q.; Berry-Kravis, E.; Kaufmann, W.E.; FORWARD Consortium. Sleep problems in fragile X syndrome: Cross-sectional analysis of a large clinic-based cohort. Am. J. Med. Genet. A 2022, 188, 1029–1039. [Google Scholar] [CrossRef]
- Tirosh, E.; Borochowitz, Z. Sleep apnea in fragile X syndrome. Am. J. Med. Genet. 1992, 43, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Niemczyk, J.; von Gontard, A.; Equit, M.; Bauer, K.; Naumann, T.; Wagner, C.; Curfs, L. Detailed assessment of incontinence in boys with fragile-X-syndrome in a home setting. Eur. J. Pediatr. 2016, 175, 1325–1334. [Google Scholar] [CrossRef]
- Carotenuto, M.; Roccella, M.; Pisani, F.; Matricardi, S.; Verrotti, A.; Farello, G.; Operto, F.F.; Bitetti, I.; Precenzano, F.; Messina, G.; et al. Polysomnographic Findings in Fragile X Syndrome Children with EEG Abnormalities. Behav. Neurol. 2019, 2019, 5202808. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Ferri, R.; Elia, M.; Del Gracco, S.; Scuderi, C.; Stefanini, M.C.; Castano, A.; Azan, G. Sleep Neurophysiology in Fragile X Patients. Dev. Brain Dysfunct. 1995, 8, 218–222. [Google Scholar]
- Miano, S.; Bruni, O.; Elia, M.; Scifo, L.; Smerieri, A.; Trovato, A.; Verrillo, E.; Terzano, M.G.; Ferri, R. Sleep phenotypes of intellectual disability: A polysomnographic evaluation in subjects with Down syndrome and Fragile-X syndrome. Clin. Neurophysiol. 2008, 119, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Balietti, M.; Casoli, T.; Di Stefano, G.; Giorgetti, B.; Aicardi, G.; Fattoretti, P. Ketogenic diets: An historical antiepileptic therapy with promising potentialities for the aging brain. Ageing Res. Rev. 2010, 9, 273–279. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- El-Rashidy, O.; El-Baz, F.; El-Gendy, Y.; Khalaf, R.; Reda, D.; Saad, K. Ketogenic diet versus gluten free casein free diet in autistic children: A case-control study. Metab. Brain Dis. 2017, 32, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Spilioti, M.; Evangeliou, A.E.; Tramma, D.; Theodoridou, Z.; Metaxas, S.; Michailidi, E.; Bonti, E.; Frysira, H.; Haidopoulou, A.; Asprangathou, D.; et al. Evidence for treatable inborn errors of metabolism in a cohort of 187 Greek patients with autism spectrum disorder (ASD). Front. Hum. Neurosci. 2013, 7, 858. [Google Scholar] [CrossRef]
- Evangeliou, A.; Vlachonikolis, I.; Mihailidou, H.; Spilioti, M.; Skarpalezou, A.; Makaronas, N.; Prokopiou, A.; Christodoulou, P.; Liapi-Adamidou, G.; Helidonis, E.; et al. Application of a ketogenic diet in children with autistic behavior: Pilot study. J. Child. Neurol. 2003, 18, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kasprowska-Liśkiewicz, D.; Liśkiewicz, A.D.; Nowacka-Chmielewska, M.M.; Nowicka, J.; Małecki, A.; Barski, J.J. The ketogenic diet affects the social behavior of young male rats. Physiol. Behav. 2017, 179, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Fortin, J.A.; Bisnauth, S.N.; Masino, S.A. Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol. Behav. 2017, 168, 138–145. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Murphy, M.I.; Slade, S.L.; Masino, S.A. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS ONE 2017, 12, e0171643. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Svedova, J.; Cote, J.L.; Sandau, U.; Rho, J.M.; Kawamura, M.; Boison, D.; Masino, S.A. Ketogenic diet improves core symptoms of autism in BTBR mice. PLoS ONE 2013, 8, e65021. [Google Scholar] [CrossRef]
- Verpeut, J.L.; DiCicco-Bloom, E.; Bello, N.T. Ketogenic diet exposure during the juvenile period increases social behaviors and forebrain neural activation in adult Engrailed 2 null mice. Physiol. Behav. 2016, 161, 90–98. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, Y.; Tomi, M.; Shin, B.C.; Thamotharan, S.; Mazarati, A.; Sankar, R.; Wang, E.A.; Cepeda, C.; Levine, M.S.; et al. Sex-Specific Life Course Changes in the Neuro-Metabolic Phenotype of Glut3 Null Heterozygous Mice: Ketogenic Diet Ameliorates Electroencephalographic Seizures and Improves Sociability. Endocrinology 2017, 158, 936–949. [Google Scholar] [CrossRef]
- Zhang, J.; Fang, Z.; Jud, C.; Vansteensel, M.J.; Kaasik, K.; Lee, C.C.; Albrecht, U.; Tamanini, F.; Meijer, J.H.; Oostra, B.A.; et al. Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am. J. Hum. Genet. 2008, 83, 43–52. [Google Scholar] [CrossRef]
- Allen, C.N. Circadian rhythms, diet, and neuronal excitability. Epilepsia 2008, 49 (Suppl. S8), 124–126. [Google Scholar] [CrossRef]
- Genzer, Y.; Dadon, M.; Burg, C.; Chapnik, N.; Froy, O. Effect of dietary fat and the circadian clock on the expression of brain-derived neurotrophic factor (BDNF). Mol. Cell Endocrinol. 2016, 430, 49–55. [Google Scholar] [CrossRef]
- Genzer, Y.; Dadon, M.; Burg, C.; Chapnik, N.; Froy, O. Ketogenic diet delays the phase of circadian rhythms and does not affect AMP-activated protein kinase (AMPK) in mouse liver. Mol. Cell Endocrinol. 2015, 417, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Metabolism: Ketogenic diet rewires circadian clock. Nat. Rev. Endocrinol. 2017, 13, 626. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Shimba, S.; Oishi, K. Ketogenic diet induces expression of the muscle circadian gene Slc25a25 via neural pathway that might be involved in muscle thermogenesis. Sci. Rep. 2017, 7, 2885. [Google Scholar] [CrossRef]
- Oike, H. Modulation of circadian clocks by nutrients and food factors. Biosci. Biotechnol. Biochem. 2017, 81, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Uchida, D.; Ohkura, N.; Doi, R.; Ishida, N.; Kadota, K.; Horie, S. Ketogenic diet disrupts the circadian clock and increases hypofibrinolytic risk by inducing expression of plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1571–1577. [Google Scholar] [CrossRef]
- Oishi, K.; Uchida, D.; Ohkura, N.; Horie, S. PPARα deficiency augments a ketogenic diet-induced circadian PAI-1 expression possibly through PPARγ activation in the liver. Biochem. Biophys. Res. Commun. 2010, 401, 313–318. [Google Scholar] [CrossRef]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5. [Google Scholar] [CrossRef]
- Fenoglio-Simeone, K.A.; Wilke, J.C.; Milligan, H.L.; Allen, C.N.; Rho, J.M.; Maganti, R.K. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia 2009, 50, 2027–2034. [Google Scholar] [CrossRef]
- Simeone, T.A.; Samson, K.K.; Matthews, S.A.; Simeone, K.A. In vivo ketogenic diet treatment attenuates pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices from epileptic Kv 1.1α knockout mice. Epilepsia 2014, 55, e44–e49. [Google Scholar] [CrossRef]
- Jain, S.V.; Glauser, T.A. Effects of epilepsy treatments on sleep architecture and daytime sleepiness: An evidence-based review of objective sleep metrics. Epilepsia 2014, 55, 26–37. [Google Scholar] [CrossRef]
- Hallböök, T.; Lundgren, J.; Rosén, I. Ketogenic diet improves sleep quality in children with therapy-resistant epilepsy. Epilepsia 2007, 48, 59–65. [Google Scholar] [CrossRef]
- Campbell, I.G. EEG recording and analysis for sleep research. Curr. Protoc. Neurosci. 2009, 49, 10.2.1–10.2.19. [Google Scholar] [CrossRef]
- Ancoli-Israel, S.; Cole, R.; Alessi, C.; Chambers, M.; Moorcroft, W.; Pollak, C.P. The role of actigraphy in the study of sleep and circadian rhythms. Sleep 2003, 26, 342–392. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child. Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Colognola, R.M.; Ferri, R.; Gigli, G.L.; Petrella, M.A.; Sanfilippo, S.; Bergonzi, P.; Tassinari, C.A. Fragile-X syndrome: A particular epileptogenic EEG pattern. Epilepsia 1988, 29, 41–47. [Google Scholar] [CrossRef]
- Pietropaolo, S.; Guilleminot, A.; Martin, B.; D’Amato, F.R.; Crusio, W.E. Genetic-background modulation of core and variable autistic-like symptoms in Fmr1 knock-out mice. PLoS ONE 2011, 6, e17073. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Knox, A.; Hervey, C. Targeted treatments for fragile X syndrome. J. Neurodev. Disord. 2011, 3, 193–210. [Google Scholar] [CrossRef]
- Peier, A.M.; McIlwain, K.L.; Kenneson, A.; Warren, S.T.; Paylor, R.; Nelson, D.L. (Over)correction of FMR1 deficiency with YAC transgenics: Behavioral and physical features. Hum. Mol. Genet. 2000, 9, 1145–1159. [Google Scholar] [CrossRef]
- Inoue, S.; Shimoda, M.; Nishinokubi, I.; Siomi, M.C.; Okamura, M.; Nakamura, A.; Kobayashi, S.; Ishida, N.; Siomi, H. A role for the Drosophila fragile X-related gene in circadian output. Curr. Biol. 2002, 12, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Sluyter, F.; de Wit, S.; Oostra, B.A.; Crusio, W.E. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 2002, 12, 39–46. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Smith, C.B. Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 15758–15763. [Google Scholar] [CrossRef]
- Dockendorff, T.C.; Su, H.S.; McBride, S.M.; Yang, Z.; Choi, C.H.; Siwicki, K.K.; Sehgal, A.; Jongens, T.A. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 2002, 34, 973–984. [Google Scholar] [CrossRef]
- Spencer, C.M.; Alekseyenko, O.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes. Brain Behav. 2005, 4, 420–430. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Smith, C.B. A null mutation for Fmr1 in female mice: Effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience 2005, 135, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Yamaguchi, T.; Hamano, K.; Siomi, H.; Saez, L.; Ishida, N.; Shimoda, M. Circadian phenotypes of Drosophila fragile x mutants in alternative genetic backgrounds. Zool. Sci. 2008, 25, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bushey, D.; Tononi, G.; Cirelli, C. The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 2009, 29, 1948–1961. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Mines, M.A.; King, M.K.; Sweatt, J.D.; Miller, C.A.; Jope, R.S. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 2010, 79, 632–646. [Google Scholar] [CrossRef]
- Baker, K.B.; Wray, S.P.; Ritter, R.; Mason, S.; Lanthorn, T.H.; Savelieva, K.V. Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes. Brain Behav. 2010, 9, 562–574. [Google Scholar] [CrossRef]
- Liu, Z.H.; Chuang, D.M.; Smith, C.B. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 2011, 14, 618–630. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Corbin, J.G.; Burns, M.P. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 2011, 33, 395–403. [Google Scholar] [CrossRef]
- Pacey, L.K.; Doss, L.; Cifelli, C.; van der Kooy, D.; Heximer, S.P.; Hampson, D.R. Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol. Cell Neurosci. 2011, 46, 563–572. [Google Scholar] [CrossRef]
- Spencer, C.M.; Alekseyenko, O.; Hamilton, S.M.; Thomas, A.M.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Modifying behavioral phenotypes in Fmr1KO mice: Genetic background differences reveal autistic-like responses. Autism Res. 2011, 4, 40–56. [Google Scholar] [CrossRef]
- Uutela, M.; Lindholm, J.; Louhivuori, V.; Wei, H.; Louhivuori, L.M.; Pertovaara, A.; Akerman, K.; Castrén, E.; Castrén, M.L. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes. Brain Behav. 2012, 11, 513–523. [Google Scholar] [CrossRef]
- Uutela, M.; Lindholm, J.; Rantamäki, T.; Umemori, J.; Hunter, K.; Võikar, V.; Castrén, M.L. Distinctive behavioral and cellular responses to fluoxetine in the mouse model for Fragile X syndrome. Front. Cell Neurosci. 2014, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, C.C.; Heitzer, A.M.; Roth, A.K.; Nawrocki, L.; Valdovinos, M.G. Effects of clonidine and methylphenidate on motor activity in Fmr1 knockout mice. Neurosci. Lett. 2015, 585, 109–113. [Google Scholar] [CrossRef]
- Sørensen, E.M.; Bertelsen, F.; Weikop, P.; Skovborg, M.M.; Banke, T.; Drasbek, K.R.; Scheel-Krüger, J. Hyperactivity and lack of social discrimination in the adolescent Fmr1 knockout mouse. Behav. Pharmacol. 2015, 26, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Saré, R.M.; Levine, M.; Smith, C.B. Behavioral Phenotype of Fmr1 Knock-Out Mice during Active Phase in an Altered Light/Dark Cycle. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.L.; Davenport, M.H.; Grainger, L.M.; Robinson, C.K.; Earnheart, A.T.; Stegman, M.S.; Lang, A.L.; Ashworth, A.A.; Molinaro, G.; Huber, K.M.; et al. Acamprosate in a mouse model of fragile X syndrome: Modulation of spontaneous cortical activity, ERK1/2 activation, locomotor behavior, and anxiety. J. Neurodev. Disord. 2017, 9, 6. [Google Scholar] [CrossRef]
- Nolan, S.O.; Reynolds, C.D.; Smith, G.D.; Holley, A.J.; Escobar, B.; Chandler, M.A.; Volquardsen, M.; Jefferson, T.; Pandian, A.; Smith, T.; et al. Deletion of. Brain Behav. 2017, 7, e00800. [Google Scholar] [CrossRef]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef] [PubMed]
- Gauducheau, M.; Lemaire-Mayo, V.; D’Amato, F.R.; Oddi, D.; Crusio, W.E.; Pietropaolo, S. Age-specific autistic-like behaviors in heterozygous Fmr1-KO female mice. Autism Res. 2017, 10, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, L.; Yin, J.; Yin, H.; Huang, Y.; Tian, J. Hyperactivity, Memory Defects, and Craniofacial Abnormalities in Zebrafish fmr1 Mutant Larvae. Behav. Genet. 2020, 50, 152–160. [Google Scholar] [CrossRef]
- Van Alphen, B.; Yap, M.H.; Kirszenblat, L.; Kottler, B.; van Swinderen, B. A dynamic deep sleep stage in Drosophila. J. Neurosci. 2013, 33, 6917–6927. [Google Scholar] [CrossRef]
- Nielsen, D.M.; Derber, W.J.; McClellan, D.A.; Crnic, L.S. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 2002, 927, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.J.; Asafo-Adjei, P.K.; Arnold, H.M.; Brown, R.E.; Bauchwitz, R.P. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes. Brain Behav. 2004, 3, 337–359. [Google Scholar] [CrossRef]
- Yan, Q.J.; Rammal, M.; Tranfaglia, M.; Bauchwitz, R.P. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005, 49, 1053–1066. [Google Scholar] [CrossRef]
- Veeraragavan, S.; Bui, N.; Perkins, J.R.; Yuva-Paylor, L.A.; Paylor, R. The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav. Neurosci. 2011, 125, 783–790. [Google Scholar] [CrossRef]
- Veeraragavan, S.; Graham, D.; Bui, N.; Yuva-Paylor, L.A.; Wess, J.; Paylor, R. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav. Brain Res. 2012, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Bui, N.; Perkins, J.R.; Yuva-Paylor, L.A.; Paylor, R. Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology 2012, 219, 47–58. [Google Scholar] [CrossRef]
- Goebel-Goody, S.M.; Wilson-Wallis, E.D.; Royston, S.; Tagliatela, S.M.; Naegele, J.R.; Lombroso, P.J. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes. Brain Behav. 2012, 11, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.; He, L.; Maaswinkel, H.; Zhu, L.; Sirotkin, H.; Weng, W. Anxiety, hyperactivity and stereotypy in a zebrafish model of fragile X syndrome and autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 55, 40–49. [Google Scholar] [CrossRef]
- Hodges, S.L.; Nolan, S.O.; Reynolds, C.D.; Lugo, J.N. Spectral and temporal properties of calls reveal deficits in ultrasonic vocalizations of adult Fmr1 knockout mice. Behav. Brain Res. 2017, 332, 50–58. [Google Scholar] [CrossRef]
- Bonasera, S.J.; Chaudoin, T.R.; Goulding, E.H.; Mittek, M.; Dunaevsky, A. Decreased home cage movement and oromotor impairments in adult Fmr1-KO mice. Genes. Brain Behav. 2017, 16, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Saré, R.M.; Harkless, L.; Levine, M.; Torossian, A.; Sheeler, C.A.; Smith, C.B. Deficient Sleep in Mouse Models of Fragile X Syndrome. Front. Mol. Neurosci. 2017, 10, 280. [Google Scholar] [CrossRef]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Byerly, M.J.; Sweeney, J.A. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry 2016, 6, e787. [Google Scholar] [CrossRef] [PubMed]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Pedapati, E.V.; Erickson, C.A.; Byerly, M.J.; Sweeney, J.A. Neural synchronization deficits linked to cortical hyper-excitability and auditory hypersensitivity in fragile X syndrome. Mol. Autism 2017, 8, 22. [Google Scholar] [CrossRef]
- Wang, J.; Ethridge, L.E.; Mosconi, M.W.; White, S.P.; Binder, D.K.; Pedapati, E.V.; Erickson, C.A.; Byerly, M.J.; Sweeney, J.A. A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J. Neurodev. Disord. 2017, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.; Featherstone, R.; Naschek, M.; Nam, J.; Du, A.; Wright, S.; Pance, K.; Melnychenko, O.; Weger, R.; Akuzawa, S.; et al. GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 knockout mouse model of fragile X syndrome. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Lovelace, J.W.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Translation-relevant EEG phenotypes in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 2018, 115, 39–48. [Google Scholar] [CrossRef]
- Wen, T.H.; Lovelace, J.W.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Developmental Changes in EEG Phenotypes in a Mouse Model of Fragile X Syndrome. Neuroscience 2019, 398, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Jonak, C.R.; Lovelace, J.W.; Ethell, I.M.; Razak, K.A.; Binder, D.K. Multielectrode array analysis of EEG biomarkers in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 2020, 138, 104794. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, J.W.; Rais, M.; Palacios, A.R.; Shuai, X.S.; Bishay, S.; Popa, O.; Pirbhoy, P.S.; Binder, D.K.; Nelson, D.L.; Ethell, I.M.; et al. Deletion of Fmr1 from Forebrain Excitatory Neurons Triggers Abnormal Cellular, EEG, and Behavioral Phenotypes in the Auditory Cortex of a Mouse Model of Fragile X Syndrome. Cereb. Cortex 2020, 30, 969–988. [Google Scholar] [CrossRef]
- Jonak, C.R.; Sandhu, M.S.; Assad, S.A.; Barbosa, J.A.; Makhija, M.; Binder, D.K. The PDE10A Inhibitor TAK-063 Reverses Sound-Evoked EEG Abnormalities in a Mouse Model of Fragile X Syndrome. Neurotherapeutics 2021, 18, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Saraf, T.S.; McGlynn, R.P.; Bhatavdekar, O.M.; Booth, R.G.; Canal, C.E. FPT, a 2-Aminotetralin, Is a Potent Serotonin 5-HT. ACS Chem. Neurosci. 2022, 13, 3629–3640. [Google Scholar] [CrossRef]
- Wong, H.; Hooper, A.W.M.; Niibori, Y.; Lee, S.J.; Hategan, L.A.; Zhang, L.; Karumuthil-Melethil, S.; Till, S.M.; Kind, P.C.; Danos, O.; et al. Sexually dimorphic patterns in electroencephalography power spectrum and autism-related behaviors in a rat model of fragile X syndrome. Neurobiol. Dis. 2020, 146, 105118. [Google Scholar] [CrossRef] [PubMed]
- Kenny, A.; Wright, D.; Stanfield, A.C. EEG as a translational biomarker and outcome measure in fragile X syndrome. Transl. Psychiatry 2022, 12, 34. [Google Scholar] [CrossRef]
- Van der Molen, M.J.; Van der Molen, M.W. Reduced alpha and exaggerated theta power during the resting-state EEG in fragile X syndrome. Biol. Psychol. 2013, 92, 216–219. [Google Scholar] [CrossRef]
- Radwan, B.; Dvorak, D.; Fenton, A.A. Impaired cognitive discrimination and discoordination of coupled theta-gamma oscillations in Fmr1 knockout mice. Neurobiol. Dis. 2016, 88, 125–138. [Google Scholar] [CrossRef]
- Arbab, T.; Battaglia, F.P.; Pennartz, C.M.A.; Bosman, C.A. Abnormal hippocampal theta and gamma hypersynchrony produces network and spike timing disturbances in the Fmr1-KO mouse model of Fragile X syndrome. Neurobiol. Dis. 2018, 114, 65–73. [Google Scholar] [CrossRef]
- Boone, C.E.; Davoudi, H.; Harrold, J.B.; Foster, D.J. Abnormal Sleep Architecture and Hippocampal Circuit Dysfunction in a Mouse Model of Fragile X Syndrome. Neuroscience 2018, 384, 275–289. [Google Scholar] [CrossRef]
- Saré, R.M.; Song, A.; Loutaev, I.; Cook, A.; Maita, I.; Lemons, A.; Sheeler, C.; Smith, C.B. Negative Effects of Chronic Rapamycin Treatment on Behavior in a Mouse Model of Fragile X Syndrome. Front. Mol. Neurosci. 2017, 10, 452. [Google Scholar] [CrossRef]
- Abolghasemi, A.; Carullo, M.P.; Aguilera, E.C.; Laroui, A.; Plantefeve, R.; Rojas, D.; Benachenhou, S.; Ramírez, M.V.; Proteau-Lemieux, M.; Lepage, J.F.; et al. Alteration of Fatty Acid Profile in Fragile X Syndrome. Int. J. Mol. Sci. 2022, 23, 10815. [Google Scholar] [CrossRef]
- Schaefer, T.L.; Ashworth, A.A.; Tiwari, D.; Tomasek, M.P.; Parkins, E.V.; White, A.R.; Snider, A.; Davenport, M.H.; Grainger, L.M.; Becker, R.A.; et al. GABA. Front. Psychiatry 2021, 12, 678090. [Google Scholar] [CrossRef] [PubMed]
- Berzhanskaya, J.; Phillips, M.A.; Gorin, A.; Lai, C.; Shen, J.; Colonnese, M.T. Disrupted Cortical State Regulation in a Rat Model of Fragile X Syndrome. Cereb. Cortex 2017, 27, 1386–1400. [Google Scholar] [CrossRef]
- Masi, D.; Spoltore, M.E.; Rossetti, R.; Watanabe, M.; Tozzi, R.; Caputi, A.; Risi, R.; Balena, A.; Gandini, O.; Mariani, S.; et al. The Influence of Ketone Bodies on Circadian Processes Regarding Appetite, Sleep and Hormone Release: A Systematic Review of the Literature. Nutrients 2022, 14, 1410. [Google Scholar] [CrossRef] [PubMed]
- Titos, I.; Juginović, A.; Vaccaro, A.; Nambara, K.; Gorelik, P.; Mazor, O.; Rogulja, D. A gut-secreted peptide suppresses arousability from sleep. Cell 2023, 186, 2273–2274. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Fujikura, Y.; Ohkura, N.; Higo-Yamamoto, S.; Mishima, T.; Oishi, K. A ketogenic diet containing medium-chain triglycerides reduces REM sleep duration without significant influence on mouse circadian phenotypes. Food Res. Int. 2023, 169, 112852. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Holst, S.C.; Sousek, A.; Hefti, K.; Saberi-Moghadam, S.; Buck, A.; Ametamey, S.M.; Scheidegger, M.; Franken, P.; Henning, A.; Seifritz, E.; et al. Cerebral mGluR5 availability contributes to elevated sleep need and behavioral adjustment after sleep deprivation. Elife 2017, 6, e28751. [Google Scholar] [CrossRef] [PubMed]
- Westmark, C.J.; Malter, J.S. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007, 5, e52. [Google Scholar] [CrossRef]
- Filon, M.J.; Wallace, E.; Wright, S.; Douglas, D.J.; Steinberg, L.I.; Verkuilen, C.L.; Westmark, P.R.; Maganti, R.K.; Westmark, C.J. Sleep and diurnal rest-activity rhythm disturbances in a mouse model of Alzheimer’s disease. Sleep 2020, 43, zsaa087. [Google Scholar] [CrossRef] [PubMed]
- Crusio, W.E. Flanking gene and genetic background problems in genetically manipulated mice. Biol. Psychiatry 2004, 56, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Crusio, W.E.; Goldowitz, D.; Holmes, A.; Wolfer, D. Standards for the publication of mouse mutant studies. Genes. Brain Behav. 2009, 8, 1–4. [Google Scholar] [CrossRef]
- Wallace, E.; Kim, D.Y.; Kim, K.M.; Chen, S.; Blair Braden, B.; Williams, J.; Jasso, K.; Garcia, A.; Rho, J.M.; Bimonte-Nelson, H.; et al. Differential effects of duration of sleep fragmentation on spatial learning and synaptic plasticity in pubertal mice. Brain Res. 2015, 1615, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.; Wright, S.; Schoenike, B.; Roopra, A.; Rho, J.M.; Maganti, R.K. Altered circadian rhythms and oscillation of clock genes and sirtuin 1 in a model of sudden unexpected death in epilepsy. Epilepsia 2018, 59, 1527–1539. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Actigraphy [4] | Sleep EEG | Concur |
|---|---|---|---|
| KD start at P18 | |||
| Genotype effects | none (AIN-76A) | WT ↑ sleep (AIN-76A Z10–12) | - |
| Fmr1KO ↑ activity (KD Z0–6) ([4] Figure 2D) | Fmr1KO ↑ wake and ↓ sleep, NREM (KD Z2–4) (Figure 3) | ✓ | |
| Fmr1KO ↑ wake and ↓ NREM (KD Z22–24) (Figure 3) | - | ||
| Diet effects | KD ↓ activity (WT and Fmr1KO Z0–6) ([4] Figure 2D) | KD ↓ REM Z0–12 (Figure 2 and Figure S5) | - |
| KD ↓ activity (WT and Fmr1KO Z12–24) ([4] Figure 2D) | KD ↑ sleep, NREM, REM (WT and Fmr1KO Z12–24) (Figure 2 and Figure S5) | ✓ | |
| KD start at 2–3 months of age | |||
| Genotype effects | Fmr1KO ↑ activity (AIN76A Z0–6) ([4] Figure 3E) | TBD | - |
| Diet effects | KD ↓ activity (Fmr1KO Z12-24) ([4] Figure 3E) | TBD | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westmark, P.R.; Gholston, A.K.; Swietlik, T.J.; Maganti, R.K.; Westmark, C.J. Ketogenic Diet Affects Sleep Architecture in C57BL/6J Wild Type and Fragile X Mice. Int. J. Mol. Sci. 2023, 24, 14460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914460
Westmark PR, Gholston AK, Swietlik TJ, Maganti RK, Westmark CJ. Ketogenic Diet Affects Sleep Architecture in C57BL/6J Wild Type and Fragile X Mice. International Journal of Molecular Sciences. 2023; 24(19):14460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914460
Chicago/Turabian StyleWestmark, Pamela R., Aaron K. Gholston, Timothy J. Swietlik, Rama K. Maganti, and Cara J. Westmark. 2023. "Ketogenic Diet Affects Sleep Architecture in C57BL/6J Wild Type and Fragile X Mice" International Journal of Molecular Sciences 24, no. 19: 14460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914460