
Proteomics Reveals mRNA Regulation and the Action of Annexins in Thyroid Cancer

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
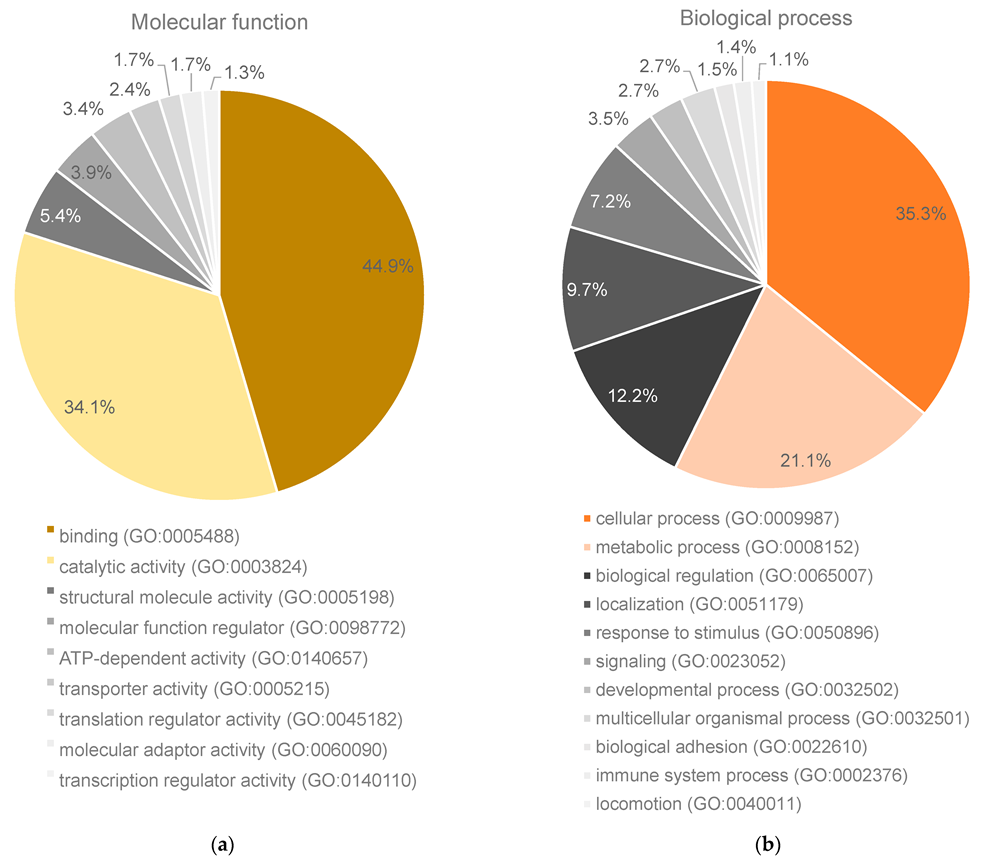
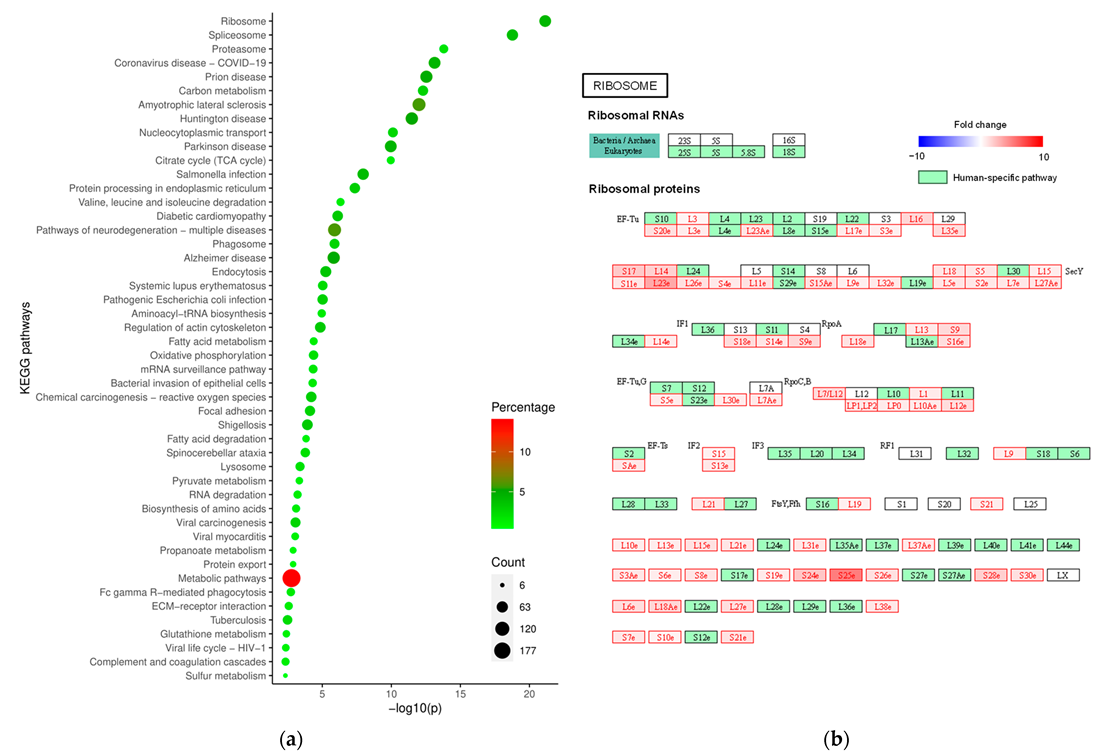
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Materials and Methods
Appendix A.2. Materials and Methods
References
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Sherman, S.I. Thyroid carcinoma. Lancet 2003, 361, 501–511. [Google Scholar] [CrossRef]
- Sipos, J.A.; Mazzaferri, E.L. Thyroid cancer epidemiology and prognostic variables. Clin. Oncol. 2010, 22, 395–404. [Google Scholar] [CrossRef]
- Damante, G.; Scaloni, A.; Tell, G. Thyroid tumors: Novel insights from proteomic studies. Expert Rev. Proteom. 2009, 6, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Jessnitzer, B.; Fuhrer, D. Proteomics in thyroid tumor research. J. Clin. Endocrinol. Metab. 2009, 94, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Pagni, F.; L’Imperio, V.; Bono, F.; Garancini, M.; Roversi, G.; De Sio, G.; Galli, M.; Smith, A.J.; Chinello, C.; Magni, F. Proteome analysis in thyroid pathology. Expert Rev. Proteom. 2015, 12, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Ucal, Y.; Ozpinar, A. Proteomics in thyroid cancer and other thyroid-related diseases: A review of the literature. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140510. [Google Scholar] [CrossRef] [PubMed]
- Sofiadis, A.; Becker, S.; Hellman, U.; Hultin-Rosenberg, L.; Dinets, A.; Hulchiy, M.; Zedenius, J.; Wallin, G.; Foukakis, T.; Hoog, A.; et al. Proteomic profiling of follicular and papillary thyroid tumors. Eur. J. Endocrinol. 2012, 166, 657–667. [Google Scholar] [CrossRef]
- Gawin, M.; Wojakowska, A.; Pietrowska, M.; Marczak, L.; Chekan, M.; Jelonek, K.; Lange, D.; Jaksik, R.; Gruca, A.; Widlak, P. Proteome profiles of different types of thyroid cancers. Mol. Cell Endocrinol. 2018, 472, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Aguilar, J.; Clifton-Bligh, R.; Molloy, M.P. Proteomics of thyroid tumours provides new insights into their molecular composition and changes associated with malignancy. Sci. Rep. 2016, 6, 23660. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, K.M.; Lindstrom, M.S. Role of ribosomal protein mutations in tumor development (Review). Int. J. Oncol. 2016, 48, 1313–1324. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, M.; Guo, Y.; Lu, X. Annexin A10 is a novel prognostic biomarker of papillary thyroid cancer. Ir. J. Med. Sci. 2021, 190, 59–65. [Google Scholar] [CrossRef]
- Petrella, A.; Festa, M.; Ercolino, S.F.; Zerilli, M.; Stassi, G.; Solito, E.; Parente, L. Annexin-1 downregulation in thyroid cancer correlates to the degree of tumor differentiation. Cancer Biol. Ther. 2006, 5, 643–647. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, W.; Li, X.; Li, H.; Li, J.; Li, H.; He, F. ANXA1 enhances tumor proliferation and migration by regulating epithelial-mesenchymal transition and IL-6/JAK2/STAT3 pathway in papillary thyroid carcinoma. J. Cancer 2021, 12, 1295–1306. [Google Scholar] [CrossRef]
- Christensen, M.V.; Hogdall, C.K.; Jochumsen, K.M.; Hogdall, E.V.S. Annexin A2 and cancer: A systematic review. Int. J. Oncol. 2018, 52, 5–18. [Google Scholar] [CrossRef]
- Qin, Y.Y.; Huang, S.N.; Chen, G.; Pang, Y.Y.; Li, X.J.; Xing, W.W.; Wei, D.M.; He, Y.; Rong, M.H.; Tang, X.Z. Clinicopathological value and underlying molecular mechanism of annexin A2 in 992 cases of thyroid carcinoma. Comput. Biol. Chem. 2020, 86, 107258. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Z.; Niu, R.; Wang, L. Crucial role of Anxa2 in cancer progression: Highlights on its novel regulatory mechanism. Cancer Biol. Med. 2019, 16, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Guo, C.; Liu, S.; Sun, M.Z. Annexin A4 and cancer. Clin. Chim. Acta 2015, 447, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, T.; Kaserer, K.; Chung, J.Y.; Bilke, S.; Krizman, D.; Knezevic, V.; Hewitt, S.M. Proteomic expression profiling of thyroid neoplasms. Proteom. Clin. Appl. 2007, 1, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Liu, S.; Guo, C.; Wang, J.; Greenaway, F.T.; Sun, M.Z. Role of annexin A6 in cancer. Oncol. Lett. 2015, 10, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liu, S.; Greenaway, F.; Sun, M.Z. Potential role of annexin A7 in cancers. Clin. Chim. Acta 2013, 423, 83–89. [Google Scholar] [CrossRef]
- Yu, S.; Bian, H.; Gao, X.; Gui, L. Annexin A9 promotes invasion and metastasis of colorectal cancer and predicts poor prognosis. Int. J. Mol. Med. 2018, 41, 2185–2192. [Google Scholar] [CrossRef]
- Hu, Z.; Kuo, W.-L.; Neve, R.M.; Gray, J.W. Annexin A9 (ANXA9) biomarker and therapeutic target in epithelial cancer. U.S. Patent 8198254B2, 26 March 2009. [Google Scholar]
- Hua, K.; Li, Y.; Zhao, Q.; Fan, L.; Tan, B.; Gu, J. Downregulation of Annexin A11 (ANXA11) Inhibits Cell Proliferation, Invasion, and Migration via the AKT/GSK-3beta Pathway in Gastric Cancer. Med. Sci. Monit. 2018, 24, 149–160. [Google Scholar] [CrossRef]
- Song, J.; Shih Ie, M.; Chan, D.W.; Zhang, Z. Suppression of annexin A11 in ovarian cancer: Implications in chemoresistance. Neoplasia 2009, 11, 605–614. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes. Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.; Gilmour, J.; Bonifer, C. The Role of the Ubiquitously Expressed Transcription Factor Sp1 in Tissue-specific Transcriptional Regulation and in Disease. Yale J. Biol. Med. 2016, 89, 513–525. [Google Scholar]
- Nicolson, N.G.; Paulsson, J.O.; Juhlin, C.C.; Carling, T.; Korah, R. Transcription Factor Profiling Identifies Spatially Heterogenous Mediators of Follicular Thyroid Cancer Invasion. Endocr. Pathol. 2020, 31, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Cao, Z.G.; Zhou, Z.W.; Han, S.J. Circ0005654 as a new biomarker of thyroid cancer interacting with SP1 to influence the prognosis: A case-control study. Medicine 2023, 102, e32853. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, S.; Shi, Y.; Chen, S. Positive feedback loop SP1/SNHG1/miR-199a-5p promotes the malignant properties of thyroid cancer. Biochem. Biophys. Res. Commun. 2020, 522, 724–730. [Google Scholar] [CrossRef]
- Ganesan, T.; Sinniah, A.; Ibrahim, Z.A.; Chik, Z.; Alshawsh, M.A. Annexin A1: A Bane or a Boon in Cancer? A Systematic Review. Molecules 2020, 25, 3700. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Griffith, K.A.; Michael, C.W.; Pu, R.T. Nodule heterogeneity as shown by size differences between the targeted nodule and the tumor in thyroidectomy specimen: A cause for a false-negative diagnosis of papillary thyroid carcinoma on fine-needle aspiration. Cancer 2008, 114, 27–33. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Galetti, V. Iodine intake as a risk factor for thyroid cancer: A comprehensive review of animal and human studies. Thyroid. Res. 2015, 8, 8. [Google Scholar] [CrossRef]
- Anjo, S.I.; Simoes, I.; Castanheira, P.; Graos, M.; Manadas, B. Use of recombinant proteins as a simple and robust normalization method for untargeted proteomics screening: Exhaustive performance assessment. Talanta 2019, 205, 120163. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Santa, C.; Manadas, B. Short GeLC-SWATH: A fast and reliable quantitative approach for proteomic screenings. Proteomics 2015, 15, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I. Current Proteomic Approaches Applied to Brain Function; Santamaría, E., Fernández-Irigoyen, J., Eds.; Springer: New York, NY, USA, 2017; pp. 107–138. [Google Scholar]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich enables enrichment analysis of OMICs datasets. J. Mol. Biol. 2020, 433, 166747. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA—Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- Ali, S.Z.; Baloch, Z.W.; Cochand-Priollet, B.; Schmitt, F.C.; Vielh, P.; VanderLaan, P.A. The 2023 Bethesda System for Reporting Thyroid Cytopathology. Thyroid 2023, 33, 1039–1044. [Google Scholar] [CrossRef]
- Lloyd, R.; Osamura, R.; Rosai, J. WHO Classification of Tumours Editorial Board. In Endocrine and Neuroendocrine Tumours; IARC: Lyon, France, 2022. [Google Scholar]
- Tang, W.H.; Shilov, I.V.; Seymour, S.L. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J. Proteome Res. 2008, 7, 3661–3667. [Google Scholar] [CrossRef] [PubMed]
- Sennels, L.; Bukowski-Wills, J.C.; Rappsilber, J. Improved results in proteomics by use of local and peptide-class specific false discovery rates. BMC Bioinform. 2009, 10, 179. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sex | Female (%) | Age (Years) | BMI (kg/m2) | Free T4 (ng/dL) | TSH (mU/L) |
|---|---|---|---|---|---|---|
| Benign | Female = 24 Male = 9 | 72.7 | 58.4 ± 2.3 | 28.7 ± 0.9 ** | 1.11 ± 0.07 | 0.83 ± 0.12 |
| Malignant | Female = 8 Male = 2 | 80.0 | 53.2 ± 7.1 | 23.7 ± 1.0 | 1.08 ± 0.04 | 1.28 ± 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, M.; Capela, J.; Anjo, S.I.; Pacheco, J.; Fernandes, M.S.; Amendoeira, I.; Jones, J.G.; Raposo, L.; Manadas, B. Proteomics Reveals mRNA Regulation and the Action of Annexins in Thyroid Cancer. Int. J. Mol. Sci. 2023, 24, 14542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914542
Coelho M, Capela J, Anjo SI, Pacheco J, Fernandes MS, Amendoeira I, Jones JG, Raposo L, Manadas B. Proteomics Reveals mRNA Regulation and the Action of Annexins in Thyroid Cancer. International Journal of Molecular Sciences. 2023; 24(19):14542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914542
Chicago/Turabian StyleCoelho, Margarida, João Capela, Sandra I. Anjo, João Pacheco, Margarida Sá Fernandes, Isabel Amendoeira, John G. Jones, Luís Raposo, and Bruno Manadas. 2023. "Proteomics Reveals mRNA Regulation and the Action of Annexins in Thyroid Cancer" International Journal of Molecular Sciences 24, no. 19: 14542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241914542