
Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches
 ,
,  , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results
2.1. A Negatively Charged Lipid Composition Results in High Load of SQV Incorporation in Liposomes
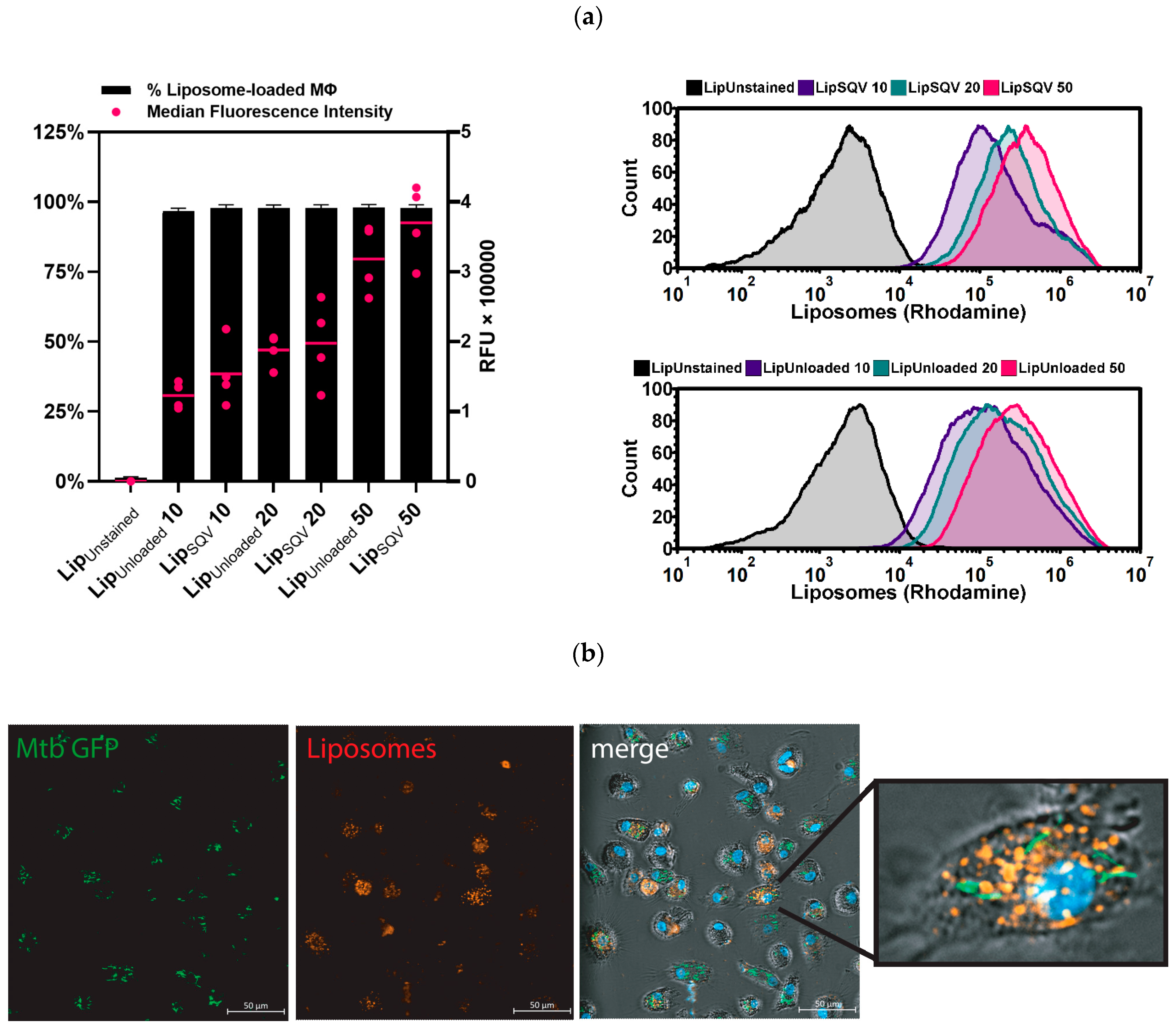
2.2. SQV-Loaded Liposomes Are Effectively Internalized by Mtb-Infected Macrophages
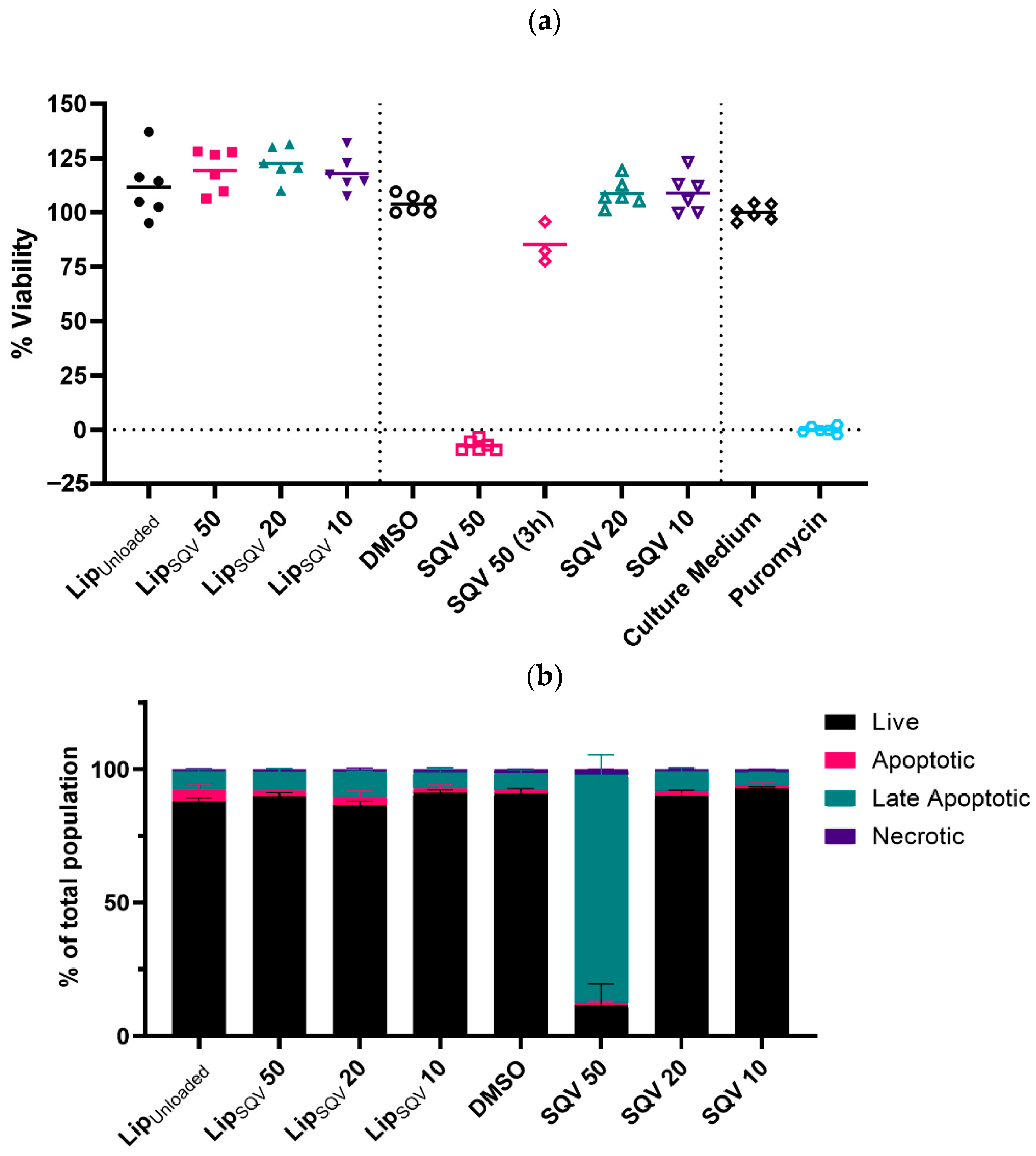
2.3. SQV-Loaded Liposomes Present No Cytotoxicity at Therapeutic Concentrations
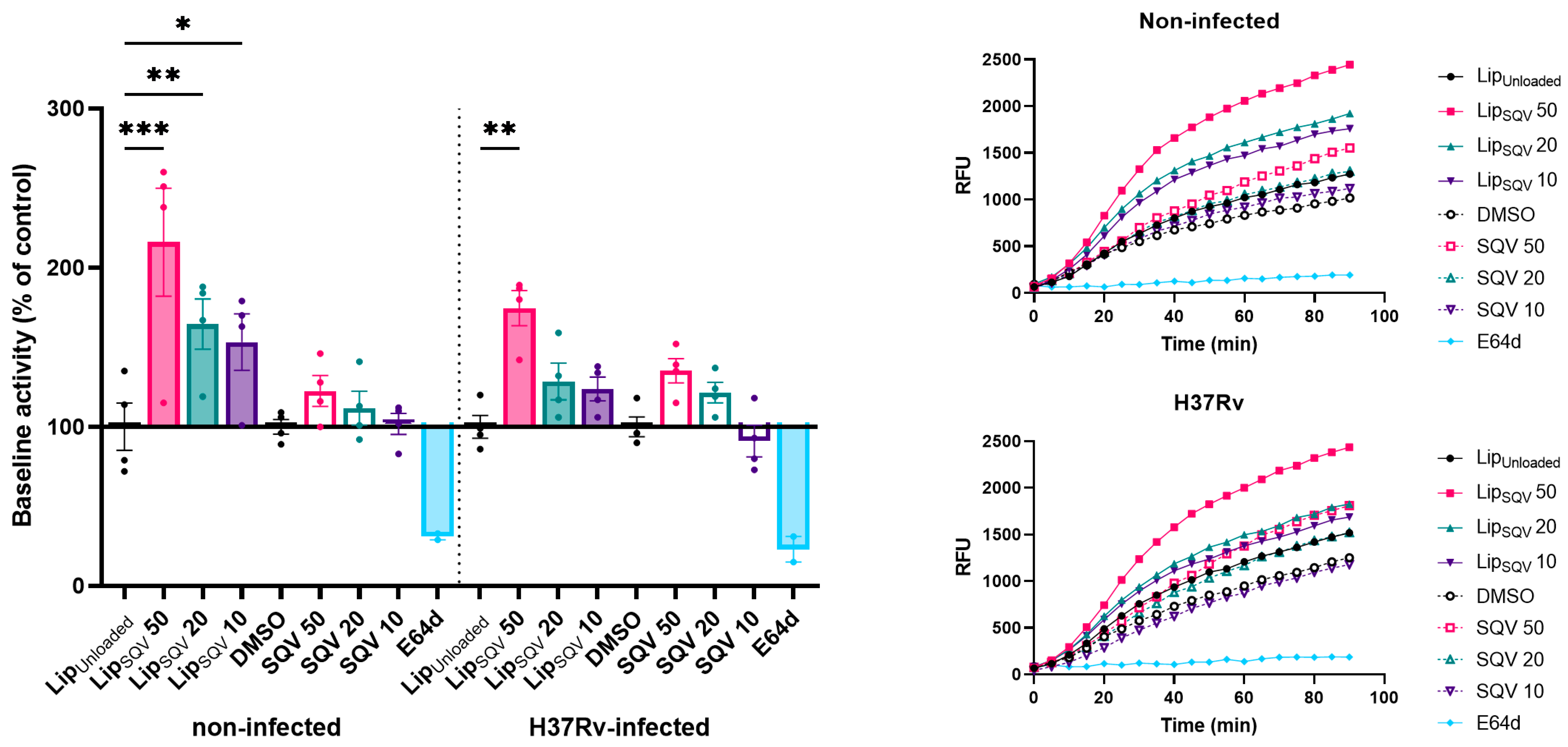
2.4. SQV-Loaded Liposomes Significantly Improve the Free-SQV Ability to Increase Cathepsin Activity
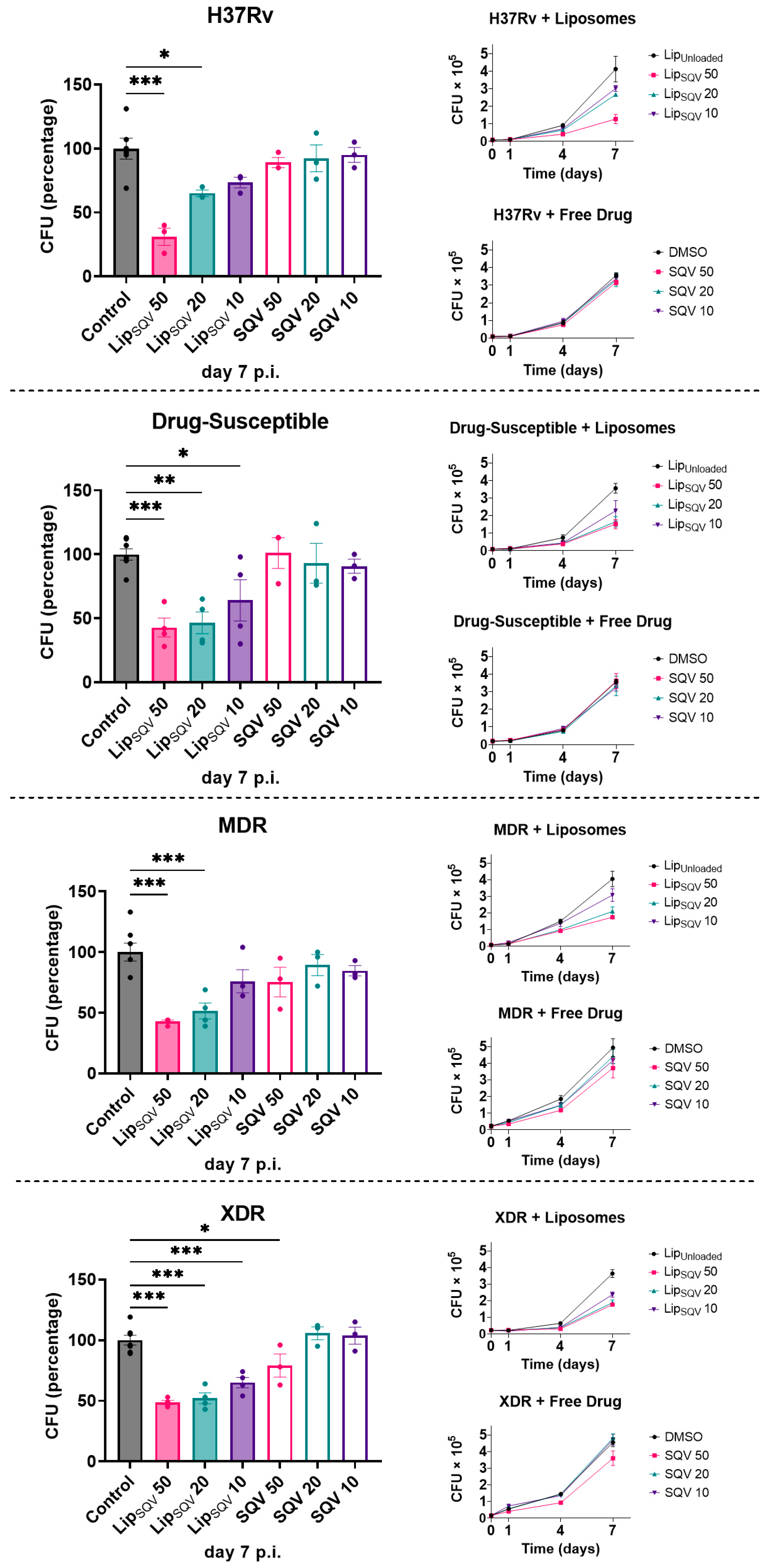
2.5. SQV-Loaded Liposomes Improve the Intracellular Killing of Mtb Reference Laboratory and Clinical Strains with Different Drug Resistance Profiles
3. Discussion
4. Materials and Methods
4.1. Preparation and Physicochemical Characterization of Saquinavir Liposomes
4.2. Cell Isolation and Culture Conditions
4.3. Bacterial Cultures
4.4. Macrophage Infection and Treatment
4.5. Flow Cytometry
4.6. Fluorescence Microscopy
4.7. Macrophage Viability
4.8. Proteolytic Activity
4.9. Bacteria Intracellular Survival
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hershberg, R.; Lipatov, M.; Small, P.M.; Sheffer, H.; Niemann, S.; Homolka, S.; Roach, J.C.; Kremer, K.; Petrov, D.A.; Feldman, M.W.; et al. High Functional Diversity in Mycobacterium tuberculosis Driven by Genetic Drift and Human Demography. PLoS Biol. 2008, 6, e311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.G. Who Puts the Tubercle in Tuberculosis? Nat. Rev. Microbiol. 2007, 5, 39–47. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; ISBN 978-92-4-006173-6.
- World Health Organization. Guidelines on the Management of Latent Tuberculosis Infection; World Health Organization: Geneva, Switzerland, 2015; ISBN 978-92-4-154890-8.
- Nakajima, H. Editorial: Tuberculosis: A Global Emergency. In World Health; World Health Organization: Geneva, Switzerland, 2013; Volume 46, p. 3. [Google Scholar]
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host Evasion and Exploitation Schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.G. New Ways to Arrest Phagosome Maturation. Nat. Cell Biol. 2007, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.A.; Hart, P.D. Phagosome-Lysosome Interactions in Cultured Macrophages Infected with Virulent Tubercle Bacilli. Reversal of the Usual Nonfusion Pattern and Observations on Bacterial Survival. J. Exp. Med. 1975, 142, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Corleis, B.; Korbel, D.; Wilson, R.; Bylund, J.; Chee, R.; Schaible, U.E. Escape of Mycobacterium tuberculosis from Oxidative Killing by Neutrophils. Cell Microbiol. 2012, 14, 1109–1121. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E.M. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [Green Version]
- Welin, A.; Raffetseder, J.; Eklund, D.; Stendahl, O.; Lerm, M. Importance of Phagosomal Functionality for Growth Restriction of Mycobacterium tuberculosis in Primary Human Macrophages. J. Innate Immun. 2011, 3, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Moura-Alves, P.; Sonawane, A.; Hacohen, N.; Griffiths, G.; Moita, L.F.; Anes, E. Mycobacterium tuberculosis Protein ESAT-6 Is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol. 2010, 12, 1046–1063. [Google Scholar] [CrossRef]
- Danilchanka, O.; Pires, D.; Anes, E.; Niederweis, M. The Mycobacterium tuberculosis Outer Membrane Channel Protein CpnT Confers Susceptibility to Toxic Molecules. Antimicrob. Agents Chemother. 2015, 59, 2328–2336. [Google Scholar] [CrossRef]
- Pires, D.; Marques, J.; Pombo, J.P.; Carmo, N.; Bettencourt, P.; Neyrolles, O.; Lugo-Villarino, G.; Anes, E. Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages. Sci. Rep. 2016, 6, 32247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.; Bernard, E.M.; Pombo, J.P.; Carmo, N.; Fialho, C.; Gutierrez, M.G.; Bettencourt, P.; Anes, E. Mycobacterium Tuberculosis Modulates MiR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front. Immunol. 2017, 8, 1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.; Calado, M.; Velez, T.; Mandal, M.; Catalão, M.J.; Neyrolles, O.; Lugo-Villarino, G.; Vérollet, C.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin C in Human Macrophages Improves Anti-Mycobacterial Immune Responses to Mycobacterium tuberculosis Infection and Coinfection With HIV. Front. Immunol. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Anes, E.; Azevedo-Pereira, J.M.; Pires, D. Cathepsins and Their Endogenous Inhibitors in Host Defense During Mycobacterium tuberculosis and HIV Infection. Front. Immunol. 2021, 12, 726984. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Hsing, L.C.; Rudensky, A.Y. The Lysosomal Cysteine Proteases in MHC Class II Antigen Presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef]
- Ha, S.-D.; Martins, A.; Khazaie, K.; Han, J.; Chan, B.M.C.; Kim, S.O. Cathepsin B Is Involved in the Trafficking of TNF-Alpha-Containing Vesicles to the Plasma Membrane in Macrophages. J. Immunol. 2008, 181, 690–697. [Google Scholar] [CrossRef] [Green Version]
- McGlinchey, R.P.; Lee, J.C. Cysteine Cathepsins Are Essential in Lysosomal Degradation of α-Synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Riese, R.J.; Mitchell, R.N.; Villadangos, J.A.; Shi, G.P.; Palmer, J.T.; Karp, E.R.; de Sanctis, G.T.; Ploegh, H.L.; Chapman, H.A. Cathepsin S Activity Regulates Antigen Presentation and Immunity. J. Clin. Investig. 1998, 101, 2351–2363. [Google Scholar] [CrossRef]
- Roberg, K.; Johansson, U.; Ollinger, K. Lysosomal Release of Cathepsin D Precedes Relocation of Cytochrome c and Loss of Mitochondrial Transmembrane Potential during Apoptosis Induced by Oxidative Stress. Free Radic. Biol. Med. 1999, 27, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. Spatial Localization of Cathepsins: Implications in Immune Activation and Resolution during Infections. Front. Immunol. 2022, 13, 955407. [Google Scholar] [CrossRef]
- Bettencourt, P.; Pires, D.; Anes, E. Immunomodulating MicroRNAs of Mycobacterial Infections. Tuberculosis 2016, 97, 1–7. [Google Scholar] [CrossRef]
- Bragman, K. Saquinavir: An HIV Proteinase Inhibitor. Adv. Exp. Med. Biol. 1996, 394, 305–317. [Google Scholar] [CrossRef]
- Kourjian, G.; Rucevic, M.; Berberich, M.J.; Dinter, J.; Wambua, D.; Boucau, J.; le Gall, S. HIV Protease Inhibitor–Induced Cathepsin Modulation Alters Antigen Processing and Cross-Presentation. J. Immunol. 2016, 196, 3595–3607. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, F.; Rivas, I.P.; Khan, M.A.; Torres Suárez, A.I. Targeting to Macrophages: Role of Physicochemical Properties of Particulate Carriers-Liposomes and Microspheres-On the Phagocytosis by Macrophages. J. Control. Release 2002, 79, 29–40. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Cruz, A.; Penha, A.F.; Reymão, J.; Sousa, A.C.; Eleutério, C.V.; Domingues, S.A.; Fraga, A.G.; Filho, A.L.; Cruz, M.E.M.; et al. Rifabutin Encapsulated in Liposomes Exhibits Increased Therapeutic Activity in a Model of Disseminated Tuberculosis. Int. J. Antimicrob. Agents 2008, 31, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, M.M.; Calado, S.; Pereira, J.; Ferronha, H.; Correia, I.; Castro, H.; Tomás, A.M.; Cruz, M.E.M. Targeted Delivery of Paromomycin in Murine Infectious Diseases through Association to Nano Lipid Systems. Nanomedicine 2015, 11, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Kourjian, G.; Xu, Y.; Mondesire-Crump, I.; Shimada, M.; Gourdain, P.; le Gall, S. Sequence-Specific Alterations of Epitope Production by HIV Protease Inhibitors. J. Immunol. 2014, 192, 3496–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajonk, F.; Himmelsbach, J.; Riess, K.; Sommer, A.; McBride, W.H. The Human Immunodeficiency Virus (HIV)-1 Protease Inhibitor Saquinavir Inhibits Proteasome Function and Causes Apoptosis and Radiosensitization in Non-HIV-Associated Human Cancer Cells. Cancer Res. 2002, 62, 5230–5235. [Google Scholar]
- Pereira, M.; Vale, N. Repurposing Alone and in Combination of the Antiviral Saquinavir with 5-Fluorouracil in Prostate and Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 12240. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.P.; Veldkamp, A.I.; Mulder, J.W.; Meenhorst, P.L.; Lange, J.M.; Beijnen, J.H.; Hoetelmans, R.M. Once-Daily Dosing of Saquinavir and Low-Dose Ritonavir in HIV-1-Infected Individuals: A Pharmacokinetic Pilot Study. AIDS 2000, 14, F103–F110. [Google Scholar] [CrossRef] [PubMed]
- McIlleron, H.; Meintjes, G.; Burman, W.J.; Maartens, G. Complications of Antiretroviral Therapy in Patients with Tuberculosis: Drug Interactions, Toxicity, and Immune Reconstitution Inflammatory Syndrome. J. Infect. Dis. 2007, 196, S63–S75. [Google Scholar] [CrossRef] [Green Version]
- Hook, G.; Jacobsen, J.S.; Grabstein, K.; Kindy, M.; Hook, V. Cathepsin B Is a New Drug Target for Traumatic Brain Injury Therapeutics: Evidence for E64d as a Promising Lead Drug Candidate. Front. Neurol. 2015, 6, 178. [Google Scholar] [CrossRef] [Green Version]
- Magister, S.; Kos, J. Cystatins in Immune System. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Kopitar-Jerala, N.N. The Role of Cystatins in Cells of the Immune System. FEBS Lett. 2006, 580, 6295–6301. [Google Scholar] [CrossRef] [Green Version]
- Baker, R. FDA Approves 3TC and Saquinavir. Food and Drug Administration. BETA 1995, 5, 9. [Google Scholar]
- Titanji, B.K.; Aasa-Chapman, M.; Pillay, D.; Jolly, C. Protease Inhibitors Effectively Block Cell-to-Cell Spread of HIV-1 between T Cells. Retrovirology 2013, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A. Rational Design of Peptide-Based HIV Proteinase Inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, J.C.; Duncan, I.B.; Hockley, D.; Grief, C.; Roberts, N.A.; Mills, J.S. Antiviral Properties of Ro 31-8959, an Inhibitor of Human Immunodeficiency Virus (HIV) Proteinase. Antivir. Res 1991, 16, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A. Expanding the Frontiers of Existing Antiviral Drugs: Possible Effects of HIV-1 Protease Inhibitors against SARS and Avian Influenza. J. Clin. Virol. 2005, 34, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, V.F.; Goulart, F.R.V.; Granato, M.Q.; Alviano, D.S.; Alviano, C.S.; Kneipp, L.F.; Santos, A.L.S. Fonsecaea pedrosoi Sclerotic Cells: Secretion of Aspartic-Type Peptidase and Susceptibility to Peptidase Inhibitors. Front. Microbiol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilho, V.V.S.; Gonçalves, K.C.S.; Rebello, K.M.; Baptista, L.P.R.; Sangenito, L.S.; Santos, H.L.C.; Branquinha, M.H.; Santos, A.L.S.; Menna-Barreto, R.F.S.; Guimarães, A.C.; et al. Docking Simulation between HIV Peptidase Inhibitors and Trypanosoma cruzi Aspartyl Peptidase. BMC Res. Notes 2018, 11, 825. [Google Scholar] [CrossRef] [Green Version]
- Nsanzabana, C.; Rosenthal, P.J. In Vitro Activity of Antiretroviral Drugs against Plasmodium falciparum. Antimicrob. Agents Chemother. 2011, 55, 5073–5077. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Updated Guidelines for Use of Rifamycins for the Treatment of Tuberculosis Among HIV-Infected Patients Taking Protease Inhibitors or Nonnucleoside Reverse Transcriptase Inhibitors. MMWR 2004, 53, 37. [Google Scholar]
- Rae, J.M.; Johnson, M.D.; Lippman, M.E.; Flockhart, D.A. Rifampin Is a Selective, Pleiotropic Inducer of Drug Metabolism Genes in Human Hepatocytes: Studies with CDNA and Oligonucleotide Expression Arrays. J. Pharmacol. Exp. Ther. 2001, 299, 849–857. [Google Scholar]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef]
- Lange, C.; Dheda, K.; Chesov, D.; Mandalakas, A.M.; Udwadia, Z.; Horsburgh, C.R. Management of Drug-Resistant Tuberculosis. Lancet 2019, 394, 953–966. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Radomska, A.; Gobbo, O.L.; Bakowsky, U.; Radomski, M.W.; Ehrhardt, C. Targeted Delivery of Transferrin-Conjugated Liposomes to an Orthotopic Model of Lung Cancer in Nude Rats. J. Aerosol. Med. Pulm. Drug Deliv. 2012, 25, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, S.I.; Dias, J.N.R.; André, A.S.; Silva, M.L.; Martins, D.; Carrapiço, B.; Castanho, M.; Carriço, J.; Cavaco, M.; Gaspar, M.M.; et al. Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening. Pharmaceutics 2021, 13, 1598. [Google Scholar] [CrossRef] [PubMed]
- Mudigonda, K.; Jukanti, R.; Apte, S.S.; Ajjala, D.R.; Shrivastava, W.; Kandikere, V.N.; Nirogi, R.V.S. HPLC Quantification of the HIV-1 Protease Inhibitor Saquinavir in Brain and Testis of Mice. Biomed. Chromatogr. 2006, 20, 1028–1032. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Lipid Composition (Molar Ratio) | (SQV /Lip)i (μg/μmol) | (SQV /Lip)f (μg/μmol) | E.E. (%) | Ø (nm) (P.I.) | ζ Pot (mV) |
|---|---|---|---|---|---|---|
| Loaded liposomes | DOPC: DOPG (8:2) | 60.9 ± 2.7 | 53.4 ± 4.9 | 90.3 ± 6.7 | 116 ± 1 (<0.1) | −23 ± 2 |
| Unloaded liposomes | DOPC: DOPG (8:2) | na | na | na | 113 ± 1 (<0.1) | −23 ± 1 |
| Liposomes labelled with rhodamine Rho-PE (0.1 mol%) | ||||||
| Loaded liposomes | DOPC: DOPG (8:2) | 42.7 ± 0.4 | 47.7 ± 0.7 | 109.1 ± 0.8 | 114 ± 1 (<0.1) | −24 ± 2 |
| Unloaded liposomes | DOPC: DOPG (8:2) | na | na | na | 111 ± 1 (<0.1) | −25 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, D.; Mandal, M.; Pinho, J.; Catalão, M.J.; Almeida, A.J.; Azevedo-Pereira, J.M.; Gaspar, M.M.; Anes, E. Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches. Int. J. Mol. Sci. 2023, 24, 1142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021142
Pires D, Mandal M, Pinho J, Catalão MJ, Almeida AJ, Azevedo-Pereira JM, Gaspar MM, Anes E. Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches. International Journal of Molecular Sciences. 2023; 24(2):1142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021142
Chicago/Turabian StylePires, David, Manoj Mandal, Jacinta Pinho, Maria João Catalão, António José Almeida, José Miguel Azevedo-Pereira, Maria Manuela Gaspar, and Elsa Anes. 2023. "Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches" International Journal of Molecular Sciences 24, no. 2: 1142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021142