
Kalkitoxin: A Potent Suppressor of Distant Breast Cancer Metastasis
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Kalkitoxin (KT) Affects Cell Viability in Breast Cancer Cell Line MDA-MB-231
2.2. KT Reduces the Motile Ability of MDA-MB-231
2.3. KT Inhibits Communication between MDA-MB-231 Cells and Active Osteoclast
2.4. KT Alters the Expression of Epithelial-Mesenchymal Transition Markers
2.5. KT Suppresses the Expression of JAK2/STAT3
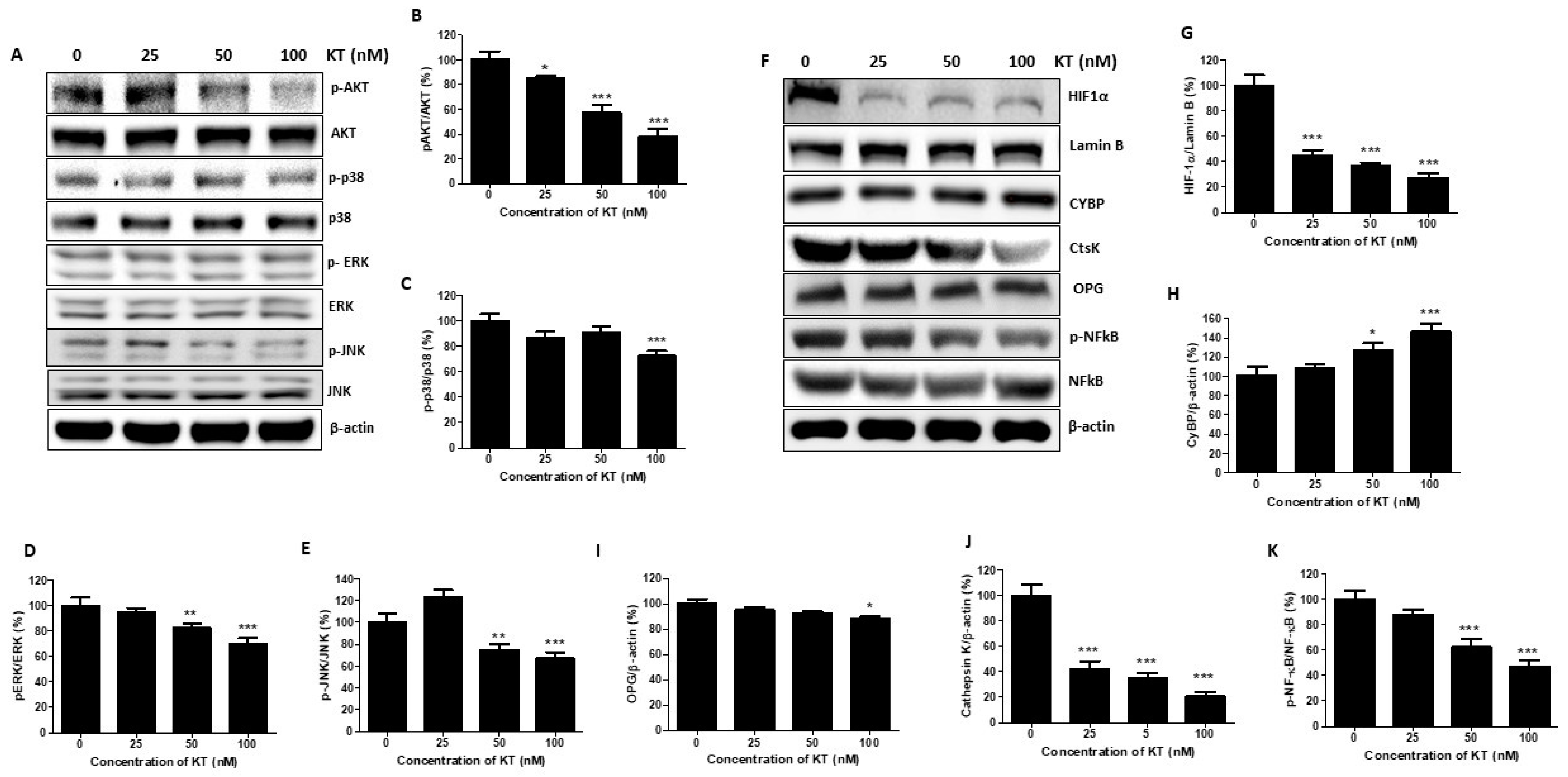
2.6. KT Inhibits the Expression of MAPK and Akt
2.7. KT Affects the Expression of Osteoclast Factors
2.8. KT Inhibits Breast Cancer Metastasis in Mouse Modeling
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell lines and Conditioned Medium
4.3. Cell Viability
4.4. Wound–Healing Assay/Transwell Migration and Invasion Assay
4.5. Preparation of Total Protein and Conditioned Media
4.6. Western Blot Analysis
4.7. Establishment of Luciferase-Expressing Stable Cells
4.8. Tumor Implantation and Drug Treatment
4.9. In Vivo Bioluminescent Imaging (BMI)
4.10. Assessment for Osteolytic Bone Metastases
4.11. Immunohistochemistry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Westbrook, J.A.; Cairns, D.A.; Peng, J.; Speirs, V.; Hanby, A.M.; Holen, I.; Wood, S.L.; Ottewell, P.; Marshall, H.; Banks, R.E.; et al. CAPG and GIPC1: Breast cancer biomarkers for bone metastasis development and treatment. J. Natl. Cancer Inst. 2016, 108, djv360. [Google Scholar] [CrossRef]
- Shi, M.; Cao, M.; Song, J.; Liu, Q.; Li, H.; Meng, F.; Pan, Z.; Bai, J.; Zheng, J. PinX1 inhibits the invasion and metastasis of human breast cancer via suppressing NF-κB/MMP-9 signaling pathway. Mol. Cancer 2015, 14, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundy, G.R. Metastasis: Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, C.L.; Moriarty, J.P.; Dusetzina, S.; Himelstein, A.L.; Foster, J.C.; Grubbs, S.S.; Novotny, P.J.; Borah, B.J. Cost-Effectiveness Analysis of Monthly Zoledronic Acid, Zoledronic Acid Every 3 Months, and Monthly Denosumab in Women With Breast Cancer and Skeletal Metastases: CALGB 70604 (Alliance). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3949–3955. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Rajasekaran, A. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behera, R.; Kumar, V.; Lohite, K.; Karnik, S.; Kundu, G.C. Activation of JAK2/STAT3 signaling by osteopontin promotes tumor growth in human breast cancer cells. Carcinogenesis 2010, 31, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Romero-Moreno, R.; Curtis, K.J.; Coughlin, T.R.; Miranda-Vergara, M.C.; Dutta, S.; Natarajan, A.; Facchine, B.A.; Jackson, K.M.; Nystrom, L.; Li, J.; et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Hurchla, M.A.; Fontana, F.; Su, X.; Amend, S.R.; Esser, A.K.; Douglas, G.J.; Mudalagiriyappa, C.; Luker, K.E.; Pluard, T.; et al. CXCR4 protein epitope mimetic antagonist POL5551 disrupts metastasis and enhances chemotherapy effect in triple-negative breast cancer. Mol. Cancer Ther. 2015, 14, 2473–2485. [Google Scholar] [CrossRef]
- Guise, T.A. Molecular mechanisms of osteolytic bone metastases. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2000, 88, 2892–2898. [Google Scholar] [CrossRef]
- Roodman, G.D.; Dougall, W. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat. Rev. 2008, 34, 92–101. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU. 1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nannini, C.J. Novel Secondary Metabolites from a Madagascar Collection of Lyngbya Majuscula. Masters Thesis, Oregon State University, Corvallis, OR, USA, 2002. [Google Scholar]
- Wu, M.; Okino, T.; Nogle, L.M.; Marquez, B.L.; Williamson, R.T.; Sitachitta, N.; Berman, F.W.; Murray, T.F.; McGough, K.; Jacobs, R.; et al. Structure, Synthesis, and Biological Properties of Kalkitoxin, a Novel Neurotoxin from the Marine Cyanobacterium Lyngbya m ajuscula. J. Am. Chem. Soc. 2000, 122, 12041–12042. [Google Scholar] [CrossRef]
- Morgan, J.B.; Liu, Y.; Coothankandaswamy, V.; Mahdi, F.; Jekabsons, M.B.; Gerwick, W.H.; Valeriote, F.A.; Zhou, Y.-D.; Nagle, D.G. Kalkitoxin inhibits angiogenesis, disrupts cellular hypoxic signaling, and blocks mitochondrial electron transport in tumor cells. Mar. Drugs 2015, 13, 1552–1568. [Google Scholar] [CrossRef] [Green Version]
- Lepage, K.T.; Goeger, D.; Yokokawa, F.; Asano, T.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. The neurotoxic lipopeptide kalkitoxin interacts with voltage-sensitive sodium channels in cerebellar granule neurons. Toxicol. Lett. 2005, 158, 133–139. [Google Scholar] [CrossRef]
- White, J.D.; Xu, Q.; Lee, C.S.; Valeriote, F.A. Total synthesis and biological evaluation of (+)-kalkitoxin, a cytotoxic metabolite of the cyanobacterium Lyngbya majuscula. Org. Biomol. Chem. 2004, 2, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.-I.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Ali, S.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Sarkar, F.H. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2012, 1826, 272–296. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal transduction pathways in breast cancer: The important role of PI3K/Akt/mTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafeiadou, V.; Hany, D.; Picard, D. Hyperactivation of MAPK induces tamoxifen resistance in SPRED2-deficient ERα-positive breast cancer. Cancers 2022, 14, 954. [Google Scholar] [CrossRef]
- Ning, X.-X.; Sun, S.-R.; Li, Y.; Gong, W.-Q.; Liu, L.-L.; Sun, L.; Liang, J.; Pan, Y.-L.; Cheng, Y.; Wu, K.-C.; et al. The effect of calcyclin binding protein on gastric cancer cell proliferation. Zhonghua Yi Xue Za Zhi 2006, 86, 3264–3268. [Google Scholar] [PubMed]
- Coniglio, S.J. Role of tumor-derived chemokines in osteolytic bone metastasis. Front. Endocrinol. 2018, 9, 313. [Google Scholar] [CrossRef]
- Roodman, D.; Silbermann, R. Mechanisms of osteolytic and osteoblastic skeletal lesions. Bonekey Rep. 2015, 4, 753. [Google Scholar]
- Langley, R.R.; Fidler, I. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; Van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; Van Diest, P.J.; Van der Wall, E. Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 1573–1581. [Google Scholar]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor–null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef] [Green Version]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; O’Higgins, N. Metalloproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Berx, G. Cadherins and Epithelial-To-Mesenchymal Transition. Prog. Mol. Biol. Transl. Sci. 2013, 116, 317–336. [Google Scholar] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Latorre, I.; Roh, M.H.; Frese, K.K.; Weiss, R.S.; Margolis, B.; Javier, R.T. Viral oncoprotein-induced mislocalization of select PDZ proteins disrupts tight junctions and causes polarity defects in epithelial cells. J. Cell Sci. 2005, 118, 4283–4293. [Google Scholar] [CrossRef] [Green Version]
- Hoevel, T.; Macek, R.; Swisshelm, K.; Kubbies, M. Reexpression of the TJ protein CLDN1 induces apoptosis in breast tumor spheroids. Int. J. Cancer 2004, 108, 374–383. [Google Scholar] [CrossRef]
- Takehara, M.; Nishimura, T.; Mima, S.; Hoshino, T.; Mizushima, T. Effect of claudin expression on paracellular permeability, migration and invasion of colonic cancer cells. Biol. Pharm. Bull. 2009, 32, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Bienz, M. β-Catenin: A pivot between cell adhesion and Wnt signalling. Curr. Biol. 2005, 15, R64–R67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.; Abbatiello, T.C.; Wu, W.L.; Kim, J.R.; Sarkissyan, M.; Sarkissyan, S.; Chung, S.S.; Elshimali, Y.; Vadgama, J.V. Slug contributes to cancer progression by direct regulation of ERα signaling pathway. Int. J. Oncol. 2015, 46, 1461–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari-Amorotti, G.; Chiodoni, C.; Shen, F.; Cattelani, S.; Soliera, A.R.; Manzotti, G.; Grisendi, G.; Dominici, M.; Rivasi, F.; Colombo, M.P.; et al. Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia 2014, 16, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Yu, H.; Tan, X.F.; Lim, T.K.; Zubaidah, R.M.; Tan, H.T.; Chung, M.C.M.; Lin, Q. Identification of key players for colorectal cancer metastasis by iTRAQ quantitative proteomics profiling of isogenic SW480 and SW620 cell lines. J. Proteome Res. 2011, 10, 4373–4387. [Google Scholar] [CrossRef]
- Le Gall, C.; Bonnelye, E.; Clézardin, P. Cathepsin K inhibitors as treatment of bone metastasis. Curr. Opin. Support Palliat. Care 2008, 2, 218–222. [Google Scholar] [CrossRef]
- Virk, M.S.; Lieberman, J.R. Tumor metastasis to bone. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S5. [Google Scholar] [CrossRef] [Green Version]
- Weichhaus, M.; Chung, S.T.M.; Connelly, L. Osteoprotegerin in breast cancer: Beyond bone remodeling. Mol. Cancer 2015, 14, 117. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Guo, W.; Ren, T.; Lou, Z.; Lu, X.; Zhang, S.; Lu, Q.; Sun, Y. Differential expression of the RANKL/RANK/OPG system is associated with bone metastasis in human non-small cell lung cancer. PLoS ONE 2013, 8, e58361. [Google Scholar] [CrossRef]
- Bhatia, P.; Sanders, M.; Hansen, M. Expression of receptor activator of nuclear factor-kappaB is inversely correlated with metastatic phenotype in breast carcinoma. Clin. Cancer Res. 2005, 11, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Azim, H.A.; Kamal, N.; Azim, H., Jr. Bone metastasis in breast cancer: The story of RANK-ligand. J. Egypt. Natl. Cancer Inst. 2012, 24, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, A.V.; Sorrels, C.; Gerwick, W. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2007, 70, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, M.; Shrestha, S.; Kim, H.; Gerwick, W.; Soh, Y. Kalkitoxin reduces osteoclast formation and resorption and protects against inflammatory bone loss. Int. J. Mol. Sci. 2021, 22, 2303. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, S.K.; Min, K.H.; Kim, S.W.; Kim, H.; Gerwick, W.H.; Soh, Y. Kalkitoxin: A Potent Suppressor of Distant Breast Cancer Metastasis. Int. J. Mol. Sci. 2023, 24, 1207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021207
Shrestha SK, Min KH, Kim SW, Kim H, Gerwick WH, Soh Y. Kalkitoxin: A Potent Suppressor of Distant Breast Cancer Metastasis. International Journal of Molecular Sciences. 2023; 24(2):1207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021207
Chicago/Turabian StyleShrestha, Saroj Kumar, Kyung Hyun Min, Se Woong Kim, Hyoungsu Kim, William H. Gerwick, and Yunjo Soh. 2023. "Kalkitoxin: A Potent Suppressor of Distant Breast Cancer Metastasis" International Journal of Molecular Sciences 24, no. 2: 1207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021207