1. Introduction
Quercetin (Que) is a 3,3′,4′,5,7-pentahydroxyflavone with the chemical formula C
15H
10O
7. The presence of five hydroxyl groups in the structure and the pyrocatechol-kind of benzene ring make Que a strong anti-oxidant and a good scavenger of free radicals [
1]. The structure of Que contains a keto carbonyl group, and the oxygen atom present on the first carbon, being basic, can form salts with acids. Furthermore, a dihydroxy group between the A ring, o-dihydroxy group B, C ring C2, C3 double bond, and 4-carbonyl are the active groups in Que (
Figure S1A). Que’s biological activities have been largely attributed to these active phenolic hydroxyl groups and double bonds [
2].
Que is highly present in daily human foods, such as onions, apples, red wine, and tea. It has various biological effects, including anti-oxidant, anti-cancer, anti-inflammatory, anti-viral, and anti-aging effects. Additionally, it has anti-aggregant and vasodilator effects, which help protect against cardiovascular diseases [
3,
4]. In humans consuming flavonoid-rich foods, the concentration range of Que plasma levels is 0.3–7.6 μM, mainly in the form of glucuronidated and sulfated metabolites [
5].
Free radicals and reactive oxygen species (ROS) are produced in cells due to common metabolic processes or external sources [
6]. Excessive oxidative stress disrupts the balance between the oxidation and anti-oxidation systems. This causes non-specific and irreversible damage to biological molecules, such as lipids, proteins, and DNA, leading to a loss of function [
7].
In many chronic diseases, such as cancer, neurodegenerative diseases, and metabolic diseases, oxidative stress is often the primary trigger [
8].
Possible sources of endogenous ROS include the nicotinamide adenine dinucleotide phosphate oxidase (NOX), xanthine oxidase (XO), lipoxygenase (LOX), myeloperoxidase (MPO), and monoamine-oxidase (MAO) enzymes [
9]. Increased activation of these enzymes has been linked to oxidative stress as well as the onset and advancement of inflammatory diseases.
Notably, Que has exhibited promising protective effects by attenuating the expression and enzymatic activity of these endogenous oxidative enzymes [
10,
11,
12,
13,
14,
15].
NOX (EC 1.6.3.1) is a transmembrane glycoprotein that catalyzes the production of superoxide (O
2−) from oxygen and NADPH. O
2− reacts quickly to produce a burst of additional oxidants, including hydrogen peroxide (H
2O
2), which is typically further converted by MPO into hypochlorous acid (HOCl) and hydroxyl radical (•OH). For this reason, NOX is usually considered a major source of ROS and oxidative stress in eukaryotic cells [
16].
XO (EC 1.17.3.2) is an iron-molybdenum flavoprotein, predominantly found in the cytoplasm of mammalian tissues, that plays a crucial role in purine catabolism [
17]. It catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid (UA) with the production of a superoxide anion. The accumulation of UA has been shown to initiate the inflammatory process through the NLRP3 inflammasome and ROS production, contributing to inflammation-related tissue damage [
18].
MPO (EC 1.11.2.2) is an enzyme found in the azurophilic/primary granules of neutrophils and, to a lesser extent, in monocytes. MPO catalyzes the oxidation of chloride and other halide ions in H
2O
2 to generate HOCl and other highly reactive products that mediate efficient anti-microbial action [
19].
LOXs (EC 1.13.11.12) are a large monomeric protein family with non-heme, non-sulfur, iron cofactor-containing dioxygenases that catalyze the oxidation of polyunsaturated fatty acids (PUFA) in lipids containing a cis,cis-1,4-pentadiene into cell signaling agents [
20]. The main products of LOXs are leukotrienes and lipoxins, which play important roles in several inflammation-related diseases, such as arthritis, asthma, cancer, and allergies.
MAO (EC 1.4.3.4) is a riboflavin protein distributed on the outer mitochondrial membrane that catalyzes the oxidative deamination of biogenic and xenobiotic amines, producing the corresponding aldehydes, hydrogen peroxide, and ammonia [
21]. Two isoforms of MAO, MAO-A and MAO-B, differ in their substrate and tissue distributions. MAO-A preferentially deaminates serotonin and norepinephrine, while MAO-B metabolizes dopamine [
22]. The generation of H
2O
2 via MAOs is reported to be a cytotoxic factor involved in oxidative stress and neurodegenerative disorders, such as Parkinson’s disease [
23].
Due to their hydrophobic nature, Que and other phenolic compounds like curcumin exhibit a strong affinity for mitochondria and can be traced through autofluorescence. This unique property of Que may contribute to its specific effect on mitochondrial dysfunction [
24].
It is also important to address the limitations of Que’s bioavailability, especially in its aglycone form, which has low solubility in water and gastrointestinal fluids, leading to precipitation and reduced absorption [
25]. Various que derivatives have differing bioavailabilities, with que glycosides found in onions having the highest absorption rates. These challenges can be overcome through strategies like encapsulation in colloidal particles and crystal engineering, as demonstrated in recent articles [
26,
27].
Our comprehensive investigation delves into Que’s potential to target oxidant enzymes implicated in ROS production. To explore these aspects thoroughly, we conducted docking analyses and molecular dynamics (MD) simulations to validate the obtained data and calculate the MMGBSA binding free energy before and during the entire dynamics. The use of docking allowed us to explore binding interactions between Que and targets, providing insight into initial structural arrangements and potential binding modes. On the other hand, the MD simulations performed provided a dynamic view of the ligand-protein complex, allowing us to assess the stability of the binding and observe any conformational changes over time. By calculating the free energy of binding with MMGBSA, we quantified the thermodynamic aspects of the ligand-protein interaction.
3. Discussion
Que is a potent anti-oxidant in the flavonoid family due to the presence of a phenolic hydroxyl group and double bonds [
40].
In recent years, several in silico studies have been conducted to elucidate the molecular basis of its anti-oxidant activity.
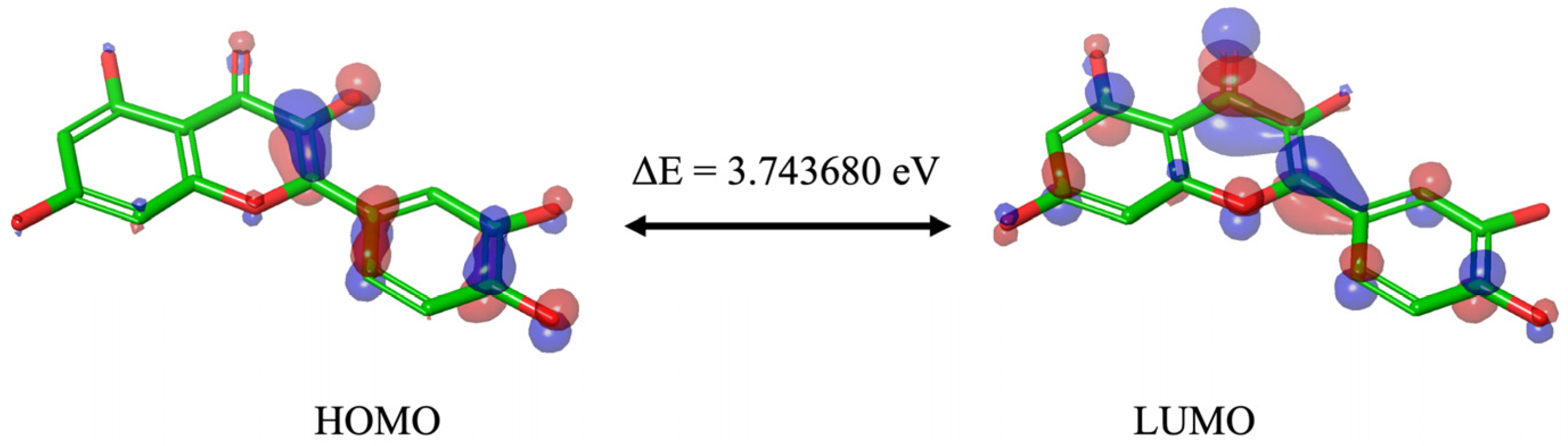
Although the different DFT studies cannot be directly compared due to the use of different software and levels of theory, the HOMO and LUMO orbitals carried out by us and Zheng et al. [
34] presented similar results, with the HOMO orbitals predominantly located in the B and C ring and the LUMO orbitals distributed throughout the molecule. Reported values are good indicators of chemical reactivity, especially for aromatic systems [
35,
36], and the results suggest that Que is a highly effective free radical scavenger.
Que anti-oxidant effects may, in part, arise from its ability to inhibit these enzymes. In fact, it has been reported that Que’s ability to attenuate the expression and enzymatic activity of various endogenous oxidative enzymes, including NOX, XO, LOX, MPO, and MAO, both in vivo and in vitro [
9,
10,
11]. To validate these observations, we conducted docking analyses and molecular dynamics simulations.
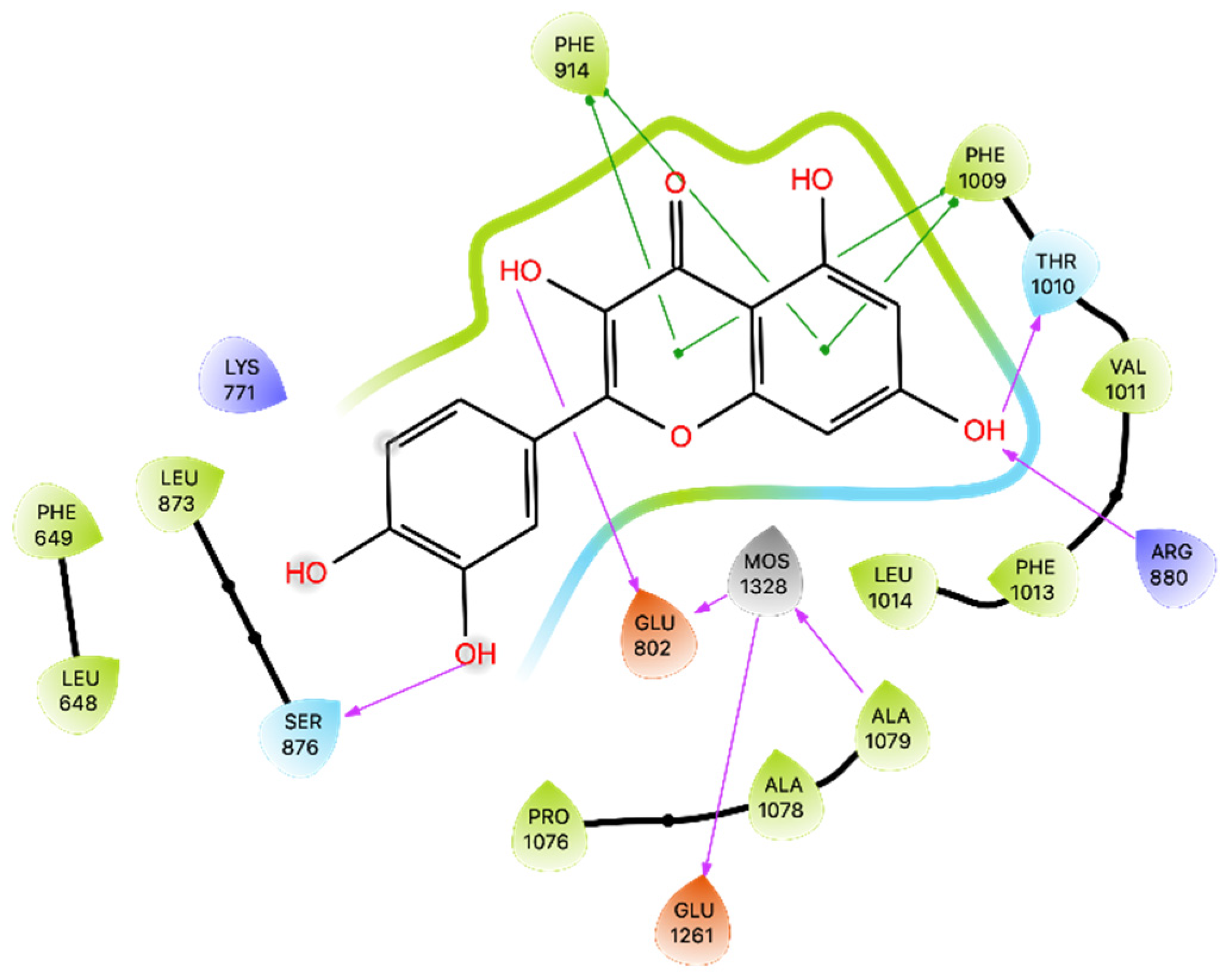
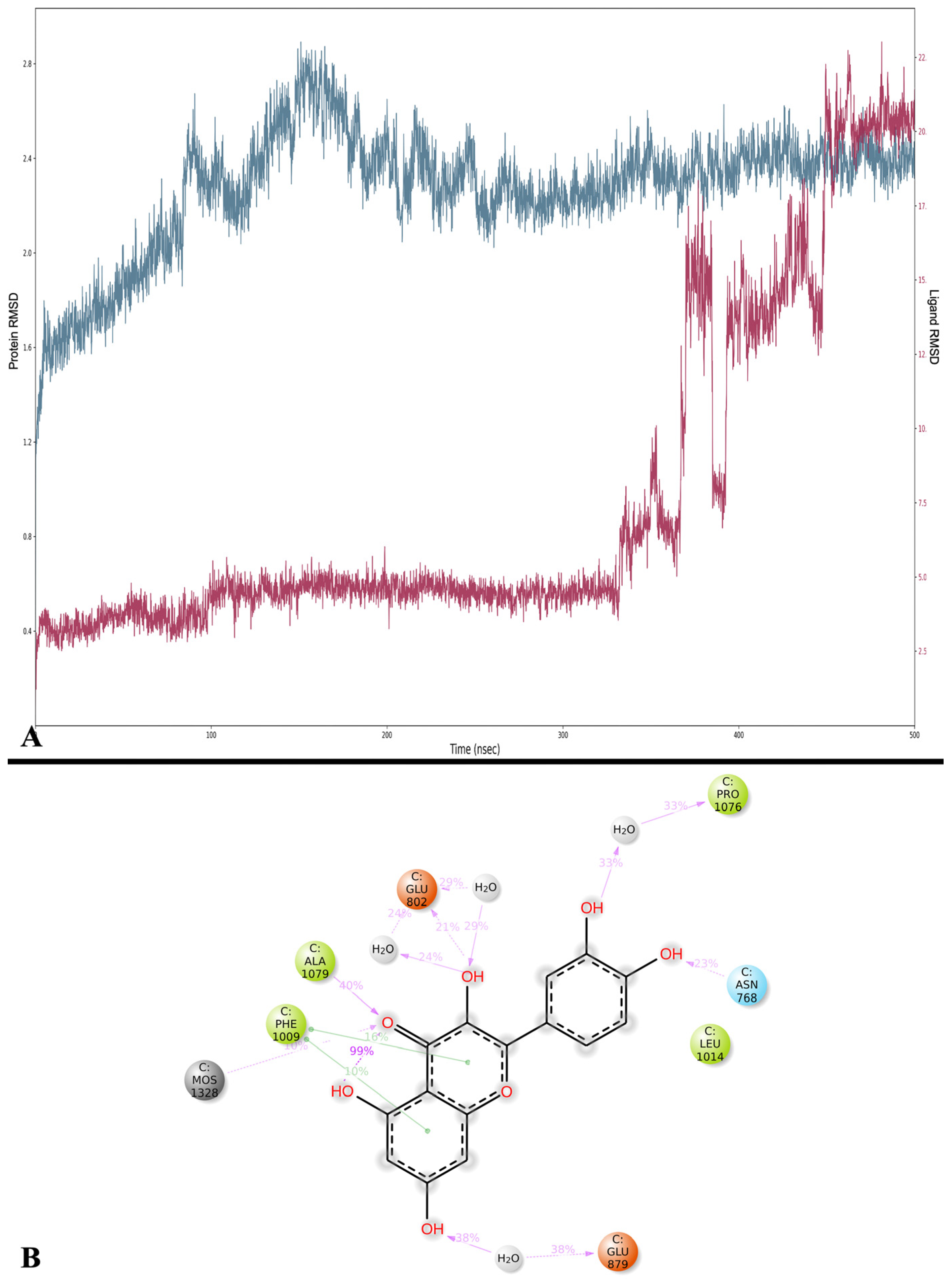
Regarding the NOX enzyme, the results of our molecular docking study raise interesting questions regarding the inhibitory potential of Que on NOX activity, especially for the MMGBSA result (−4.82 kcal/mol). These results warrant further investigation into the dynamics and binding kinetics of Que within the NOX binding pocket. The MD confirms that the binding between Que and NOX finds excellent stability after approximately 100 ns and remains stable throughout the remainder of the simulation. The new MMGBSA calculations also improved by obtaining an average of −56.421 kcal/mol. The difference is probably due to the change in binding compared to docking and the better fit of Que on NOX observed during the simulation, which is evident from the greater formation of strong interactions for most of the simulation (
Table S4).
The previous studies, including X-ray structural analysis [
37] and various docking simulations [
38,
41], consistently highlighted Que’s potential as an XO inhibitor. Building upon these promising insights, our own docking studies reaffirmed the previous findings supporting Que’s potential as an XO inhibitor. Moreover, MMGBSA energy in the docking pose yielded a substantial binding energy of −22.18 kcal/mol, indicating a discrete binding affinity confirmed by MD. The five most populated clusters also maintained binding in the same docking zone, and Que maintained excellent stability and some interactions for most of the simulation (
Table S4), losing it slightly but then regaining it.
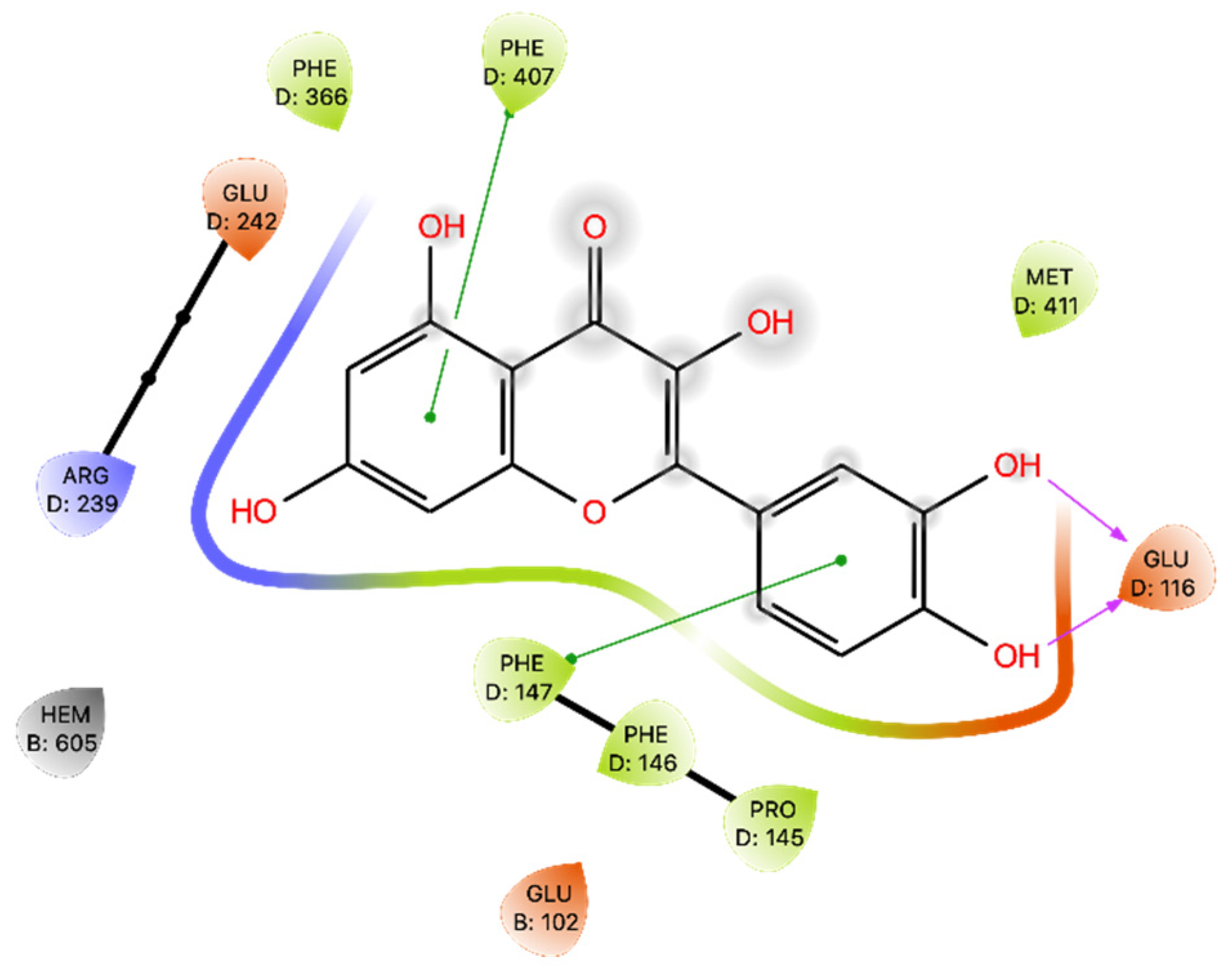
Regarding MPO, our results reaffirm the potential inhibitory role of Que in modulating MPO activity, as previously studied by Pereira and colleagues [
42]. Que effectively interacts with MPO, primarily through H bonds with Glu 116. The Glide Gscore energy values were not optimal, while the MMGBSA of the docking pose yielded a moderate energy result. The MD study revealed changes in Que binding within the binding site, as evidenced by the ligand’s RMSD exhibiting a wide range, especially at the beginning of the dynamics, where Que undergoes modifications in its binding. This is further confirmed by the range of MMGBSA values obtained, ranging from −62.350 to −14.895 kcal/mol, and by the different binding poses found in the five most populated clusters.
Regarding lipoxygenase (5-LOX) enzymes, our results are like the docking results presented by Vyshnevska et al. [
43]. The MD studies confirm the binding stability between Que and this enzyme with low RMSDs for enzyme and ligand, and the MMGBSA studies performed throughout the dynamics are comparable to docking. Furthermore, the five most populated clusters have bindings comparable to docking. These results contribute to the growing body of evidence supporting Que’s role as a potential natural agent for controlling inflammation and related disorders through 5-LOX inhibition.
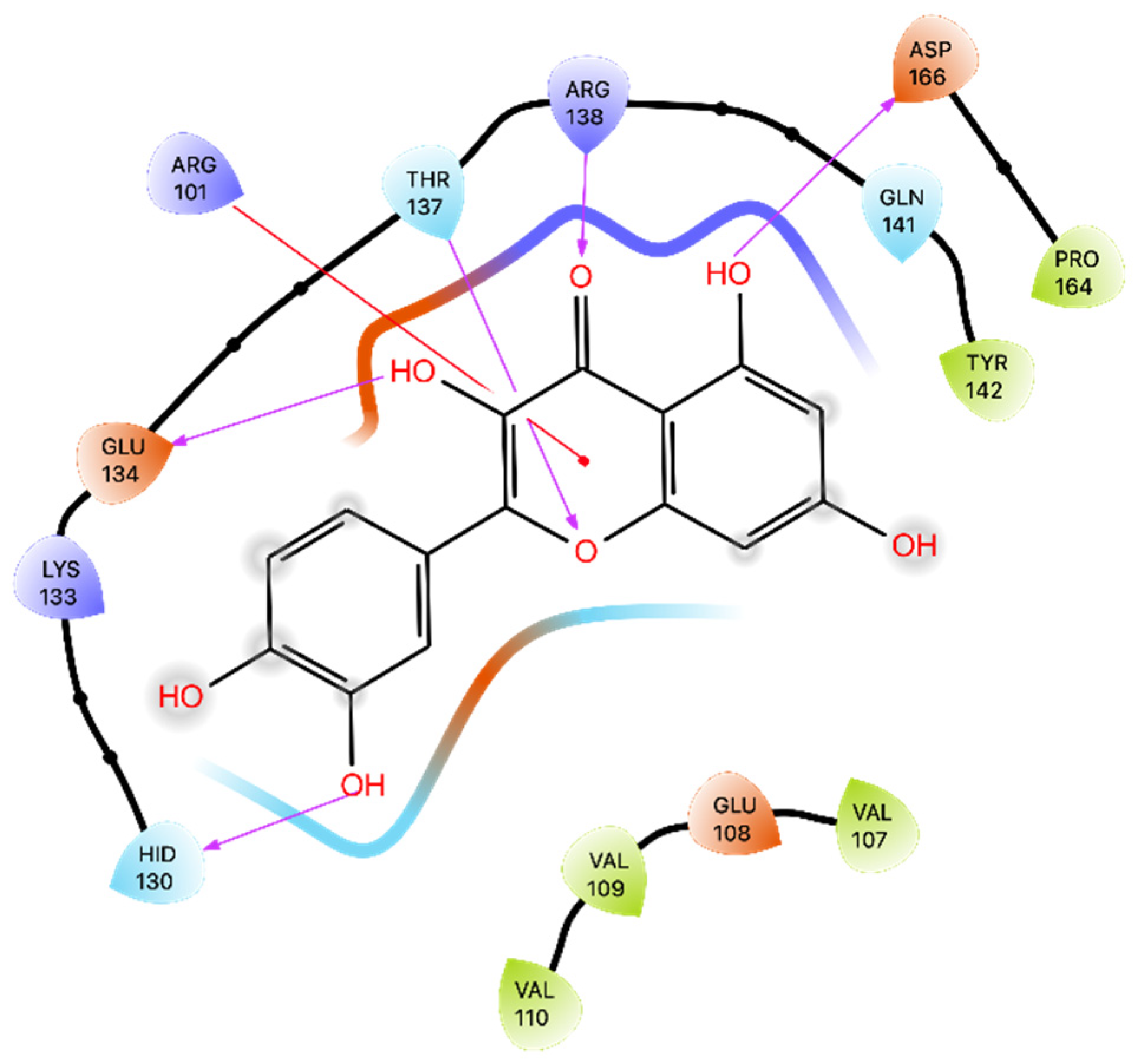
Comparing our results to those reported by Larit et al. [
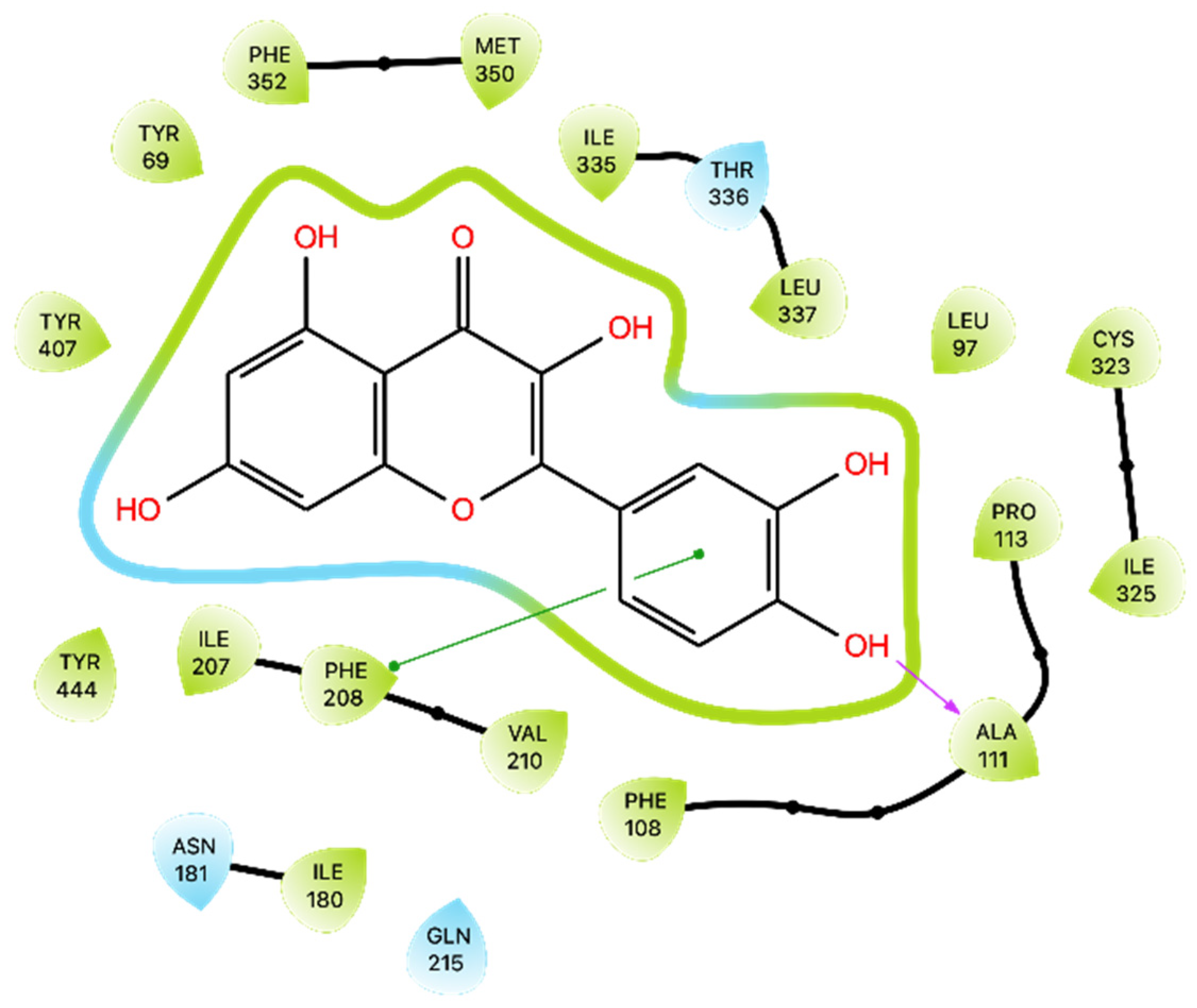
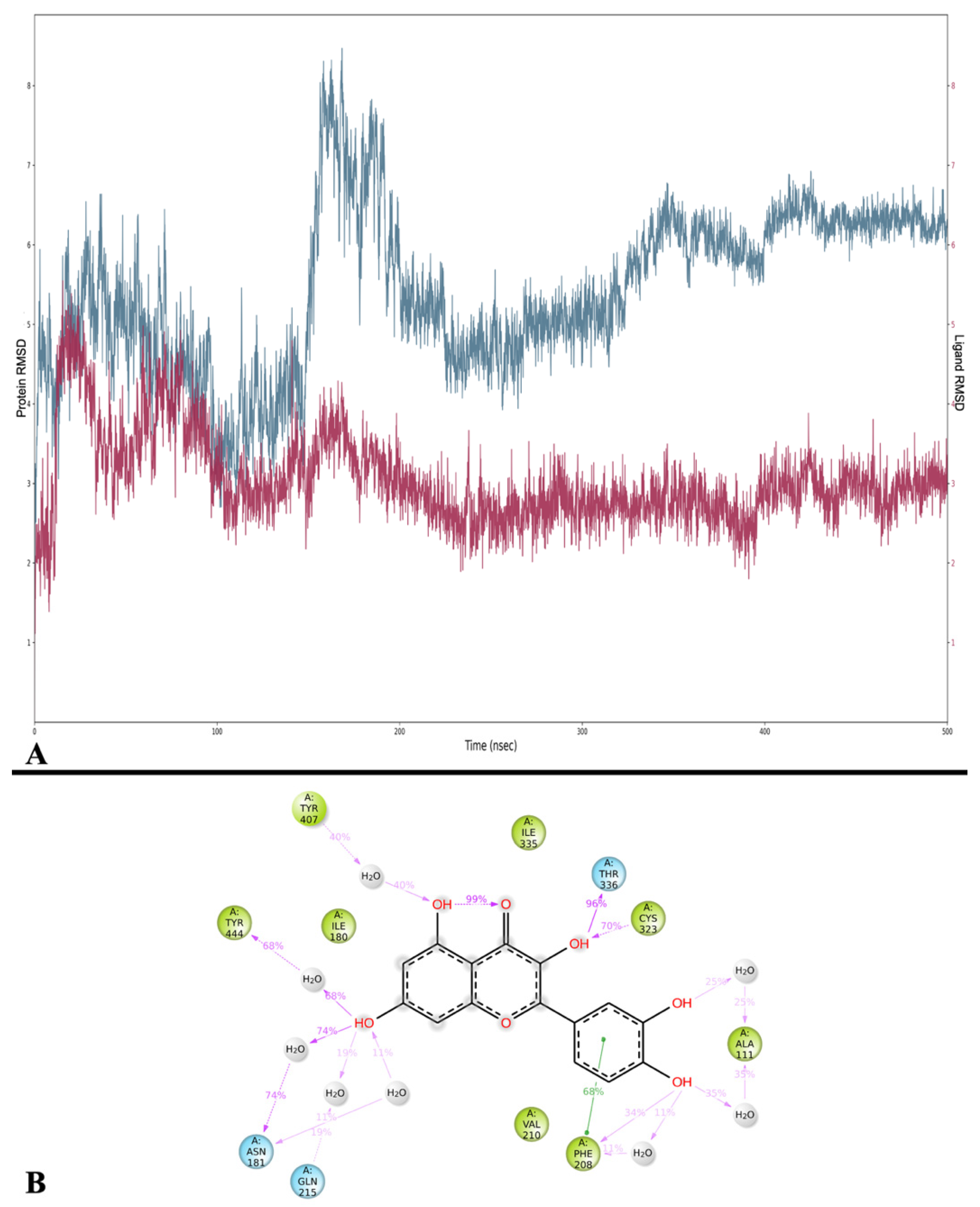
44], we observed some differences in Que’s interactions with MAO-A. Larit et al. reported that Que interacted with multiple residues, including Ala 111, Ile 180, Asn 181, Phe 208, Gln 215, Thr 336, and Tyr 444. In contrast, our docking study on the MAO-A active site showed that Que primarily formed an H-bond with Ala 111 and a π-π interaction with Phe 208. An anomalous result was found for the energy of MMGBSA for docking. MD studies overturned this positive value and obtained an average MMGBSA energy of −38.836 kcal/mol. Dynamics analysis showed that Que stabilizes after approximately 180 ns of simulation and then maintains its stability. The interactions with Ala 111 and Phe 208 are also preserved during MD, additionally forming those with Thr 336, Asn181, Cys 323, and Tyr 444. Overall, these results suggest that Que has the potential to inhibit MAO-A, albeit with variations in the specific residues involved.
5. Conclusions
Que, a flavonoid widely distributed in fruits and vegetables, is known for its anti-microbial, anti-viral, anti-oxidant, and anti-inflammatory properties. Its anti-oxidant activity is mainly attributed to its intrinsic ability to neutralize free radicals due to the hydroxyl substitutions and the catechol-type B-ring. Additionally, it can inhibit the expression of pro-inflammatory and oxidant genes and endogenous oxidizing enzymes. The results of docking studies show that Que can interact with oxidative enzymes, making it a promising candidate for developing new anti-inflammatory agents. The study found that Que demonstrated the greatest effectiveness in molecular docking simulations with XO, followed by MAO-A, 5-LOX, NOX, and MPO, which are implicated in several inflammatory pathologies. To confirm these findings and explore the mechanisms of action further, MD simulation studies were conducted over a 500 ns period. These simulations confirmed binding and contributed to a more detailed understanding of the interactions between Que and the five oxidative enzymes. For the NOX and MPO simulations, clear variations in binding with Que were observed, while excellent binding energy values calculated with MMGBSA were observed for all simulations. In addition, all enzymes except MPO showed strong interactions preserved during the simulations. In fact, the only stable and weak interaction with MPO was with Glu116 (19%). These findings collectively emphasize the remarkable potential of Que as a versatile compound with significant implications for its development as a novel class of anti-inflammatory agents, provided strategies to enhance its bioavailability are explored and implemented.
However, it is crucial to acknowledge that Que has its limitations, particularly in terms of bioavailability. Its aglycone form exhibits low systemic exposure. This is an important consideration for the potential therapeutic use of Que. Novel delivery methods, including inclusion complexes, prodrugs, nanocrystals, microemulsions, liposomes, and phospholipid formulations, as well as polymer nanoparticles and micelles, hold significant promise and warrant further exploration to improve que’s bioavailability, medicinal value, and applicability.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}