
Behçet’s Disease: A Comprehensive Review on the Role of HLA-B*51, Antigen Presentation, and Inflammatory Cascade

Abstract
:1. Introduction
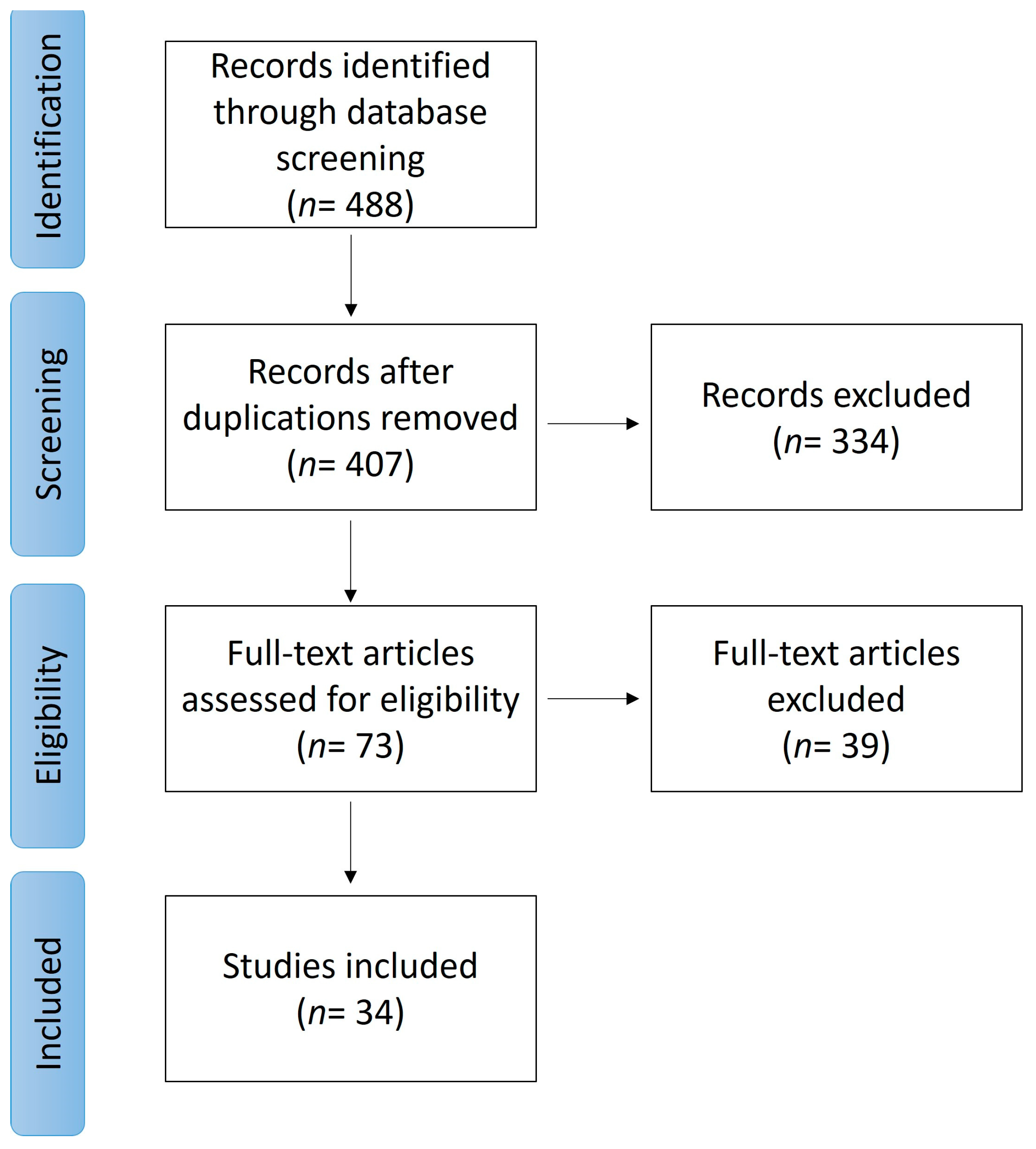
2. Methods
3. Results
3.1. HLA-B*51:01 Association: The Strongest Genetic Risk Factor

{kind=link}
{kind=link}
{kind=link}
| Reference | Study | Ethnicity | Patients | Results |
|---|---|---|---|---|
| Kim et al., 2018 [31] | R | Korean | OA: 433 BD: 126 | In HLA-B*51+ BD patients (n = 40), clinical features of the diagnostic criteria were dominant In HLA-B*27+ BD patients (n = 17), genital ulcers and skin lesions were dominant |
| Krause et al., 1999 [29] | R | Israeli | BD: 55 | HLA-B5+ patients have enhanced occurrence of thrombophlebitis, less erythema nodosum, older disease onset age, and more severe disease |
| Mizuki et al., 2020 [28] | R | Japanese | BD: 3044 | HLA-B*51+ BD patients have: Increased risk of ocular lesion (OR 1.59, 95% CI: 1.37–1.84; p < 0.001) Decreased risk of genital ulceration (OR 0.72, 95% CI: 0.62–0.84; p < 0.001) and gastrointestinal symptoms (OR 0.65, 95% CI: 0.55–0.77; p < 0.001) |
| Pamukcu et al., 2022 [27] | R | Turkish | BD: 204 | HLA-B*51+ BD patients have a higher risk of PPL (OR 1.946, 95% CI: 1.044–3.629) and ocular (OR 2.399, 95% CI: 1.165–4.938) and neurological involvement (OR 5.404, 95% CI: 1.119–26.093) |
| Rajaei et al., 2020 [18] | R | Iranian | BD: 63 | The percentage of HLA-B5 (25%) and HLA-B*51 (21%) |
| Ryu et al., 2018 [26] | R | Korean | BD: 193 | HLA-B*51+ patients: Earlier disease onset (28.3 ± 11.4 years vs. 33.8 ± 11.6 years, p = 0.02), More frequent neurologic (17.2% vs. 2.5%, p = 0.02) and gastrointestinal involvements (20.7% vs. 2.5%, p = 0.01) |
| Ideguchi et al., 2011 [17] | R | Japanese | BD: 412 | HLA-B*51 was positive in 50% (53% in male patients, 48% in female patients) Higher frequency of HLA-B*51 in patients with ocular involvement |
| Kirino et al., 2016 [30] | R | Japanese | BD: 578 | Phenotypical evolution in Japanese BD patients during the last 30 years: Significant decrease in complete-type BD, HLA-B*51 carriers, and gastrointestinal symptoms |
| Soejima et al., 2021 [32] | R | Japanese | BD: 657 (1990–2018) BD: 6754 (2003–2014) | Temporary alteration of clinical cluster proportions over time caused increasing GI involvement, reduced incidence of complete type according to Japanese criteria, and reduced HLA-B*51-positive BD patients |
| Ortiz-Fernández et al., 2016 [33] | CC | Spanish | BD: 278 HC: 1517 | Highest association with BD: HLA-B*51 (p = 6.82 × 10-32, OR 3.82) HLA-B57 (p = 1.02 × 10-5, OR 2.80, 95% CI = 1.77–4.43) and HLA-A03 (p = 9.68 × 10-3, OR 0.61, 95% CI = 0.41–0.89) identified as additional HLA genes associated with BD |
| Al-Okaily et al., 2016 [34] | CC | Saudi | BD: 60 HC: 60 | Enhanced frequency of HLA-A*26, -A*31, and -B*51 alleles in BD patients HLA-B*15 allele may have a protective effect on BD |
| Alpsoy et al., 1998 [16] | CC | Turkish | BD: 71 HC: 600 | HLA-B*51 significantly increased in BD patients DR7 significantly decreased in BD patients |
| Demirseren et al., 2014 [35] | CC | Turkish | BD: 51 HC: 44 | HLA-B*51 is significantly higher in BD patients HLA-B*5101, HLA-B*5102(01), HLA-B*5109, and HLA-B*5122 subtypes increased in BD patients Negative correlation between PPL involvement and HLA-B*5109 HLA-B*5103 may be a risk factor for neuro-Behçet |
| Hamzaoui et al., 2012 [36] | CC | Tunisian | BD: 178 HC: 125 | Higher HLAB-51 frequency in BD patients (47.19% vs. 20.8%, p < 0.001). HLA B51+ patients have: Higher frequency of pathergy test positivity (p = 0.01) Retinal vasculitis (p = 0.045) Lower frequency of arterial aneurysms (p = 0.009) and neurological involvement |
| Itoh et al., 2006 [21] | CC | Japanese | BD: 180 HC: 170 | Strong association of HLA-B*5101 with BD (Pc = 1 × 1016, OR = 8.5) Weak association between A2602 (Pc = 0.130, OR = 4.3) and HLA-B3901 (Pc = 0.099, OR = 3.5) |
| Koumantaki et al., 1998 [20] | CC | Greek | BD: 62 HC: 87 | Higher frequency of HLA-B*5101 in BD patients (80% vs. 26%) (OR 10.48, p < 10−6) Males carrying B5101 allele have a higher risk for BD compared to females (OR 16.97 vs. 5.74, respectively) |
| Mizuki et al., 2001 [37] | CC | Jordanian | BD: 49 HC: 50 | The strongest risk factor: HLA-B*51 |
| Castillo-Palma, et al., 1996 [38] | CC | Spanish | BD: 67 HC: 223 | HLA-B*51+ is higher in males with ocular (p = 0.0001), cutaneous (p = 0.001), and digestive involvement (p = 0.05) DQB1*0303 was linked to worse prognosis in uveitis (p = 0.01) DR11 and DQB1*0301 were more common in HLA B51+ patients |
| Muñoz et al., 2020 [39] | CC | Argentinian | BD: 34 HC: 240 | BD was associated with HLA-B*51 allele (OR = 3.75; p = 0.0012). |
| Pirim et al., 2004 [22] | CC | Turkish | BD: 75 HC: 54 | HLA-B*51 frequency is higher in BD (58.7% vs. 18.5%, OR = 6.245) Most prevalent class II HLA in BD were HLA-DRB1*04 (45.3%) and HLA-DRB1*07 (24%) |
| Rodríguez et al., 1998 [40] | CC | Spanish | BD: 21 HC: 25 | HLA-B*51 frequency is higher in BD (37.5% vs. 15.5%) BD patients had higher frequencies of B*5101 (32% vs. 13%), and 5108 (5.5% vs. 1.2%) |
| Sakly et al., 2009 [41] | CC | Tunisian | AS&BD: 365 HC: 124 | No significant difference in HLA-B*51 between BD and HC (30.0% vs. 16.1%, p > 0.05) |
| Sanz et al., 1998 [42] | CC | Spanish | BD: 56 HC: 66 | Association of Cw*1602 with BD (OR 20.15, corrected ρ < 0.05) with higher relative risk compared to association of B51 in this study (OR 1.85) |
| Zouboulis et al., 1993 [25] | CC | German | BD: 39 HC: 1415 | Higher frequency of HLA-B5 allele among BD patients (p < 0.05) Higher frequency of HLA-B5 in male patients with severe vascular involvement |
| Ombrello et al., 2014 [43] | CC | Turkish | BD: 1190 HC: 1257 | HLA-B*51, -A*03, -B*15, -B*27, -B*49, -B*57, and -A*26 with risk of BD Independent associations between BD and the HLA-B/MICA region and the area between HLA-F and HLA-A (p < 1.7 × 10−8) |
| Capittini et al., 2021 [44] | Ma | Worldwide | NA | The most frequent HLA-B*51 two-digit alleles associated with BD differed among populations: Europe, HLA-B*5108 (OR 11.25 C.I. 4.9–26) Turkey, HLA-B*5101 (OR 5.98 C.I. 3.7–9.8) Japan, HLA-B*5102 (OR 5.39 C.I. 0.6–47) HLA class I alleles associated with risk for BD are B*5108, B*51, B*5101, B*5102, DQB1-03, A*2601, Cw14, Cw15, Cw16, B*15, and A*26 |
| de Menthon et al., 2009 [19] | Ma | Worldwide | BD:4800 HC: 16289 | Enhanced risk of BD 5.78 and 5.9 times over in carriers of HLA–B*51/B*5 and HLA–B*51, respectively |
| Maldini et al., 2012 [24] | Ma | Worldwide | NA | HLA-B*51/B5+ positivity is associated with: Male gender Increased frequencies of genital ulcers and ophthalmic and skin manifestations Reduced frequency of gastrointestinal involvement |
| Horie et al., 2017 [23] | Ma | Worldwide | NA | HLA-B*51 strongly associated with BD ocular manifestations in East and Middle Eurasian regions (OR = 2.40, p = 0.0030 and 1.87, p = 0.0045, respectively), but not in West Eurasian regions (p = 0.35) |
| Kirino et al., 2013 [45] | GWAS | Turkish | BD: 1209 HC: 1278 | ERAP1 variants preferentially conferred risk for BD in HLA-B*51-positive individuals (p-value = 0.0009) ERAP1 p.Arg725Gln homozygosity was related with BD with an odds ratio of 3.78 [95% CI 1.94–7.35] in HLA-B*51-positive people and 1.48 [95% CI 0.78–2.80] in negative individuals New susceptibility loci detected at CCR1, STAT4, and KLRC4 for BD |
| Remmers et al., 2010 [13] | GWAS | Turkish | BD: 1214 HC: 1278 | Association of BD and HLA-B*51 (OR = 3.49, 95% CI = 2.95 to 4.12, p = 5.47 × 10−50) Identification of a second, independent association within the MHC class I region telomeric to HLA-B Association at IL10 (rs1518111, p = 1.88 × 10−8) |
| Su et al., 2022 [46] | GWAS | Chinese | BD: 1015 HC: 4502 | Association of HLA–B*51, HLA–A*26, and HLA–C*0704 with BD-related uveitis 22 new susceptibility variations in 16 non-HLA loci (RHOH, PRDM1, MTHFD1L, KLF4, ZMIZ1, RPS6KA4-PRDX5, SIPA1-FIBP-FOSL1, IL10RA, VAMP1, AGBL1, CMIP, CDH15-ZNF778, TCF4, MRPL39-JAM2, GART, and MIS18A) |
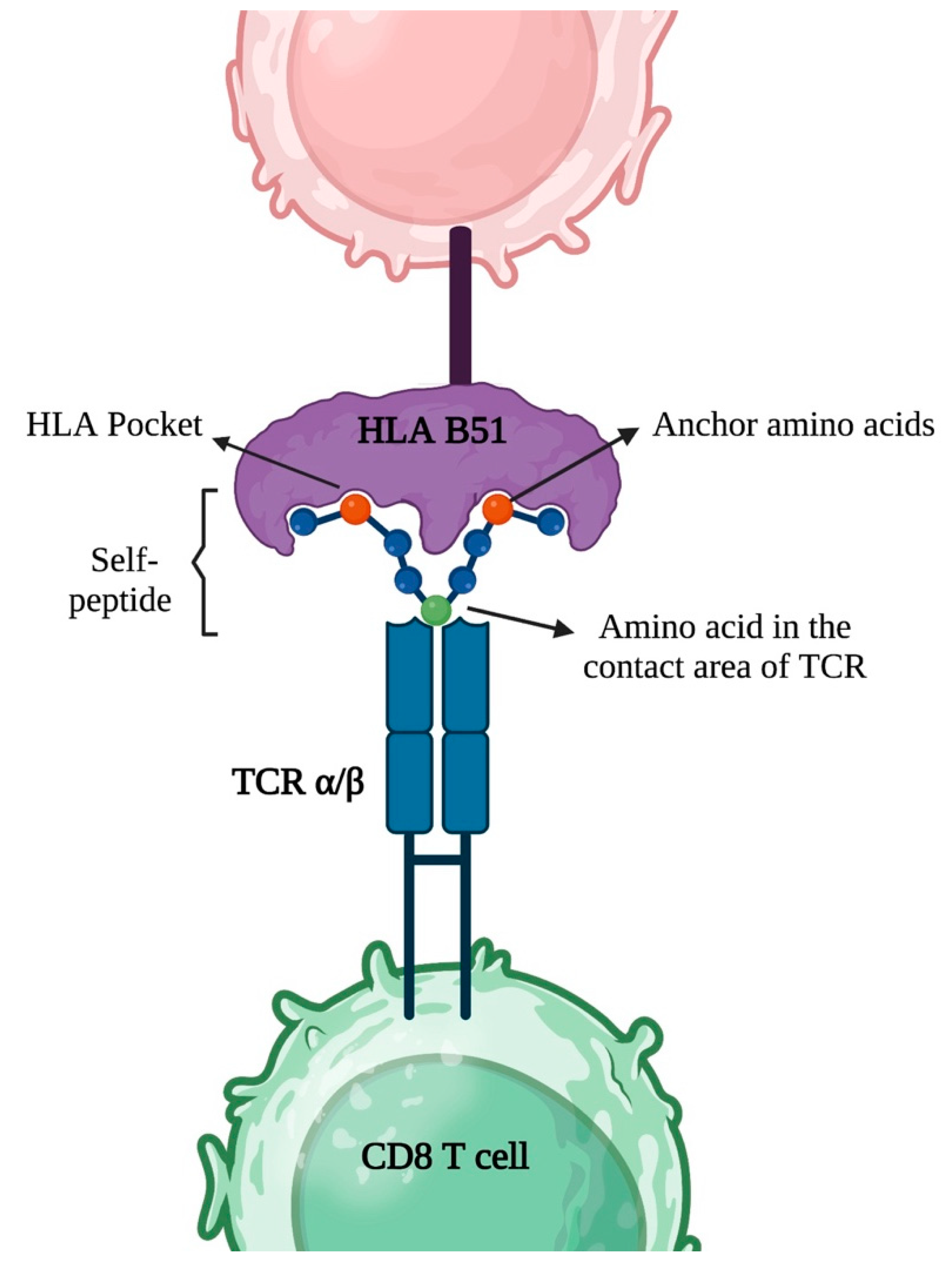
3.2. HLA Class I Association in Antigen Presentation, TCR Antigen Recognition and Inflammation
3.3. Gene Loci Related to Antigen Presentation
3.3.1. HLA-B*51 and ERAP1 Epistasis in Behçet’s Disease
3.3.2. The Repertoire of HLA-B*51 and Binding of Peptides
3.3.3. Other HLA Class I Alleles
3.3.4. Association of KIR3D Receptor Polymorphisms
3.4. Gene Loci Related to IL23/T17 Pathways
IL23R-IL12RB2 Locus
3.5. Gene Loci Related to Innate Immunity
3.5.1. Association of MEFV Gene
3.5.2. Association of FUT2 Gene
3.5.3. Toll-Like Receptor Genes
3.5.4. TNFAIP3
3.5.5. STAT4
3.5.6. Other Associated Genes
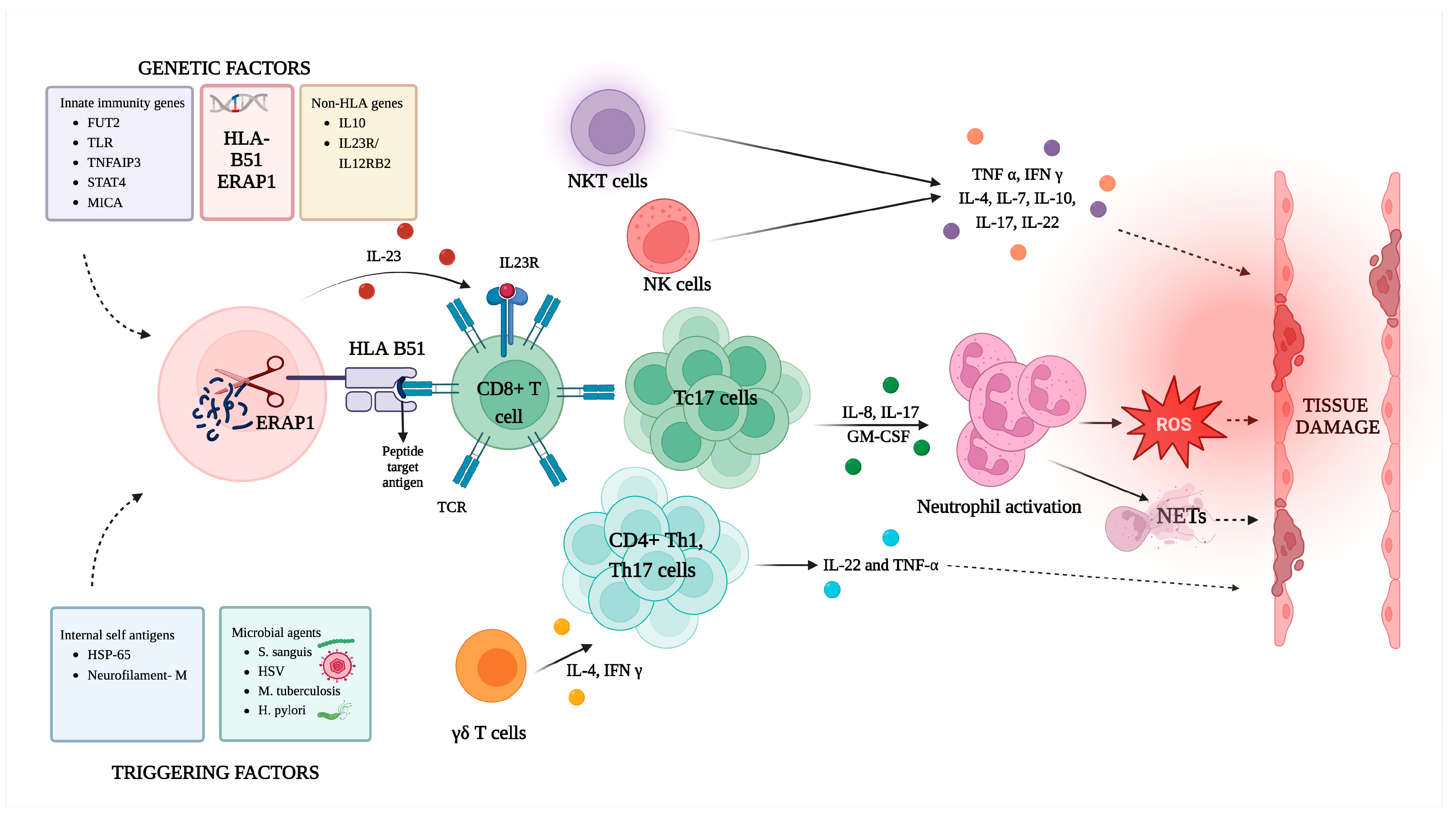
3.6. Antigens in Behçet’s Disease
3.7. Exploring the Involvement of Diverse Immune Cells in Behçet’s Disease
3.7.1. Cytotoxic T Cells (CD8+ T Cells)
3.7.2. T Helper 1 Cells
3.7.3. T Helper 17 Cells
3.7.4. Natural Killer T Cells
3.7.5. Gamma-Delta (γδ) T Cells
3.7.6. Regulatory T (Treg) Cells
3.7.7. Natural Killer Cells
- CD56brightCD16 NK cells, primarily found in the lymph nodes, specialize in cytokine release.
3.7.8. Neutrophils
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonmez, C.; Yucel, A.A.; Yesil, T.H.; Kucuk, H.; Sezgin, B.; Mercan, R.; Yucel, A.E.; Demirel, G.Y. Correlation between IL-17A/F, IL-23, IL-35 and IL-12/-23 (p40) levels in peripheral blood lymphocyte cultures and disease activity in Behcet’s patients. Clin. Rheumatol. 2018, 37, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.G.; Ortiz-Fernandez, L.; Coit, P.; Yilmaz, V.; Yentur, S.P.; Alibaz-Oner, F.; Aksu, K.; Erken, E.; Duzgun, N.; Keser, G.; et al. Sex-specific analysis in Behcet’s disease reveals higher genetic risk in male patients. J. Autoimmun. 2022, 132, 102882. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, E. Behçet’s disease: A comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J. Dermatol. 2016, 43, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Leccese, P.; Alpsoy, E. Behçet’s disease: An overview of etiopathogenesis. Front. Immunol. 2019, 10, 1067. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Boyvat, A. The skin in Behçet’s disease: Mucocutaneous findings and differential diagnosis. JEADV Clin. Pract. 2022, 1, 11–20. [Google Scholar] [CrossRef]
- Greco, A.; De Virgilio, A.; Ralli, M.; Ciofalo, A.; Mancini, P.; Attanasio, G.; de Vincentiis, M.; Lambiase, A. Behcet’s disease: New insights into pathophysiology, clinical features and treatment options. Autoimmun. Rev. 2018, 17, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Gholijani, N.; Daryabor, G.; Yazdani, M.R.; Vazani, N.; Shabbooei, B.; Zahed, M.; Ranjbar, M.A.; Sadeghi, M.B.; Amirghofran, Z. Serum interleukin-37 (IL-37) and its gene polymorphism in Iranian Behcet’s disease patients: Association with disease manifestations and activity. Meta Gene 2020, 26, 100794. [Google Scholar] [CrossRef]
- Giza, M.; Koftori, D.; Chen, L.; Bowness, P. Is Behçet’s disease a ‘class 1-opathy’? The role of HLA-B*51 in the pathogenesis of Behçet’s disease. Clin. Exp. Immunol. 2018, 191, 11–18. [Google Scholar] [CrossRef]
- Takeuchi, M.; Kastner, D.L.; Remmers, E.F. The immunogenetics of Behçet’s disease: A comprehensive review. J. Autoimmun. 2015, 64, 137–148. [Google Scholar] [CrossRef]
- Ahn, H.S.; Kim, H.J.; Kazmi, S.Z.; Kang, T.; Jun, J.B.; Kang, M.J.; Kim, K.B.; Kee, S.H.; Kim, D.S.; Hann, H.J. Familial risk of Behcet’s disease among first-degree relatives: A population-based aggregation study in Korea. Rheumatology 2021, 60, 2697–2705. [Google Scholar] [CrossRef]
- van der Houwen, T.; van Laar, J. Behçet’s disease, and the role of TNF-α and TNF-α blockers. Int. J. Mol. Sci. 2020, 21, 3072. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Aslani, S.; Meguro, A.; Akhtari, M.; Fatahi, Y.; Mizuki, N.; Shahram, F. A comprehensive overview on the genetics of Behcet’s disease. Int. Rev. Immunol. 2022, 41, 84–106. [Google Scholar] [CrossRef] [PubMed]
- Remmers, E.F.; Cosan, F.; Kirino, Y.; Ombrello, M.J.; Abaci, N.; Satorius, C.; Le, J.M.; Yang, B.; Korman, B.D.; Cakiris, A.; et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat. Genet. 2010, 42, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, S.R.; Yildirim, R.; Yazici, Y. Behcet’s Syndrome. Drugs 2012, 72, 2223–2241. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Stephen, S.; Ashchyan, H.J.; James, W.D.; Micheletti, R.G.; Rosenbach, M. Neutrophilic dermatoses: Pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J. Am. Acad. Dermatol. 2018, 79, 987–1006. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, E.; Yilmaz, E.; Coşkun, M.; Savaş, A.; Yeğin, O. HLA antigens and linkage disequilibrium patterns in Turkish Behçet’s patients. J. Dermatol. 1998, 25, 158–162. [Google Scholar] [CrossRef]
- Ideguchi, H.; Suda, A.; Takeno, M.; Ueda, A.; Ohno, S.; Ishigatsubo, Y. Behçet disease: Evolution of clinical manifestations. Medicine 2011, 90, 125–132. [Google Scholar] [CrossRef]
- Rajaei, E.; Jalali, M.T.; Pezeshki, S.M.S.; Rezaeeyan, H.; Maniati, M.; Elyasi, M.; Zayeri, Z.D. Dose HLA-B5, 7, 8, 27, and 51 Antigens Associated to Behcet’s disease? A Study in Southwestern Iran. Curr. Rheumatol. Rev. 2020, 16, 120–124. [Google Scholar] [CrossRef]
- de Menthon, M.; Lavalley, M.P.; Maldini, C.; Guillevin, L.; Mahr, A. HLA-B51/B5 and the risk of Behcet’s disease: A systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009, 61, 1287–1296. [Google Scholar] [CrossRef]
- Koumantaki, Y.; Stavropoulos, C.; Spyropoulou, M.; Messini, H.; Papademetropoulos, M.; Giziaki, E.; Marcomichelakis, N.; Palimeris, G.; Kaklamanis, P.; Kaklamani, E. HLA-B*5101 in Greek patients with Behcet’s disease. Hum. Immunol. 1998, 59, 250–255. [Google Scholar] [CrossRef]
- Itoh, Y.; Inoko, H.; Kulski, J.; Sasaki, S.; Meguro, A.; Takiyama, N.; Nishida, T.; Yuasa, T.; Ohno, S.; Mizuki, N. Four-digit allele genotyping of the HLA-A and HLA-B genes in Japanese patients with Behcet’s disease by a PCR-SSOP-Luminex method. Tissue Antigens 2006, 67, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Pirim, I.; Atasoy, M.; Ikbal, M.; Erdem, T.; Aliagaoglu, C. HLA class I and class II genotyping in patients with Behcet’s disease: A regional study of eastern part of Turkey. Tissue Antigens 2004, 64, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Meguro, A.; Ohta, T.; Lee, E.B.; Namba, K.; Mizuuchi, K.; Iwata, D.; Mizuki, N.; Ota, M.; Inoko, H.; et al. HLA-B51 Carriers are Susceptible to Ocular Symptoms of Behçet Disease and the Association between the Two Becomes Stronger towards the East along the Silk Road: A Literature Survey. Ocul. Immunol. Inflamm. 2017, 25, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Maldini, C.; Lavalley, M.P.; Cheminant, M.; de Menthon, M.; Mahr, A. Relationships of HLA-B51 or B5 genotype with Behcet’s disease clinical characteristics: Systematic review and meta-analyses of observational studies. Rheumatology 2012, 51, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.; Büttner, P.; Djawari, D.; Kirch, W.; Keitel, W.; Garbe, C.; von Keyserlingk-Eberius, H.; Orfanos, C. The HLA pattern in Adamantiades-Behcet’s disease in Germany. Association of occurrence, clinical symptoms and follow-up in 39 patients. Der Hautarzt Z. Fur Dermatol. Venerol. Und Verwandte Geb. 1993, 44, 81–85. [Google Scholar]
- Ryu, H.J.; Seo, M.R.; Choi, H.J.; Baek, H.J. Clinical phenotypes of Korean patients with Behcet disease according to gender, age at onset, and HLA-B51. Korean J. Intern. Med. 2018, 33, 1025. [Google Scholar] [CrossRef]
- Pamukcu, M.; Duran, T.I.; Demirag, M.D. HLA-B51 Impact on Clinical Symptoms in Behcet’s Disease. J. Coll. Physicians Surg. Pak. 2022, 32, 904. [Google Scholar]
- Mizuki, Y.; Horita, N.; Horie, Y.; Takeuchi, M.; Ishido, T.; Mizuki, R.; Kawagoe, T.; Shibuya, E.; Yuda, K.; Ishido, M. The influence of HLA-B51 on clinical manifestations among Japanese patients with Behcet’s disease: A nationwide survey. Mod. Rheumatol. 2020, 30, 708–714. [Google Scholar] [CrossRef]
- Krause, I.; Molad, Y.; Weinberger, A. Association of HLA-B5 with Clinical Expression and Severity of Behcet’s Disease in Israel. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 1999, 5, 137–140. [Google Scholar]
- Kirino, Y.; Ideguchi, H.; Takeno, M.; Suda, A.; Higashitani, K.; Kunishita, Y.; Takase-Minegishi, K.; Tamura, M.; Watanabe, T.; Asami, Y.; et al. Continuous evolution of clinical phenotype in 578 Japanese patients with Behçet’s disease: A retrospective observational study. Arthritis Res. Ther. 2016, 18, 217. [Google Scholar] [CrossRef]
- Kim, J.; Jung, J.; Choi, S.; Lee, Y.; Ji, J.; Song, G. SAT0530 Clinical Features Association with hla-b Allelic Types (B27, B51) in Korean Patients of Behcet’s Disease; BMJ Publishing Group Ltd.: London, UK, 2018. [Google Scholar]
- Soejima, Y.; Kirino, Y.; Takeno, M.; Kurosawa, M.; Takeuchi, M.; Yoshimi, R.; Sugiyama, Y.; Ohno, S.; Asami, Y.; Sekiguchi, A.; et al. Changes in the proportion of clinical clusters contribute to the phenotypic evolution of Behçet’s disease in Japan. Arthritis Res. Ther. 2021, 23, 49. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Fernández, L.; Carmona, F.D.; Montes-Cano, M.A.; García-Lozano, J.R.; Conde-Jaldón, M.; Ortego-Centeno, N.; Castillo, M.J.; Espinosa, G.; Graña-Gil, G.; Sánchez-Bursón, J.; et al. Genetic Analysis with the Immunochip Platform in Behçet Disease. Identification of Residues Associated in the HLA Class I Region and New Susceptibility Loci. PLoS ONE 2016, 11, e0161305. [Google Scholar] [CrossRef] [PubMed]
- Al-Okaily, F.; Al-Rashidi, S.; Al-Balawi, M.; Mustafa, M.; Arfin, M.; Al-Asmari, A. Genetic association of HLA-A*26,-A*31, and-B*51 with Behcet’s disease in Saudi patients. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2016, 9, CMAMD-S39879. [Google Scholar] [CrossRef] [PubMed]
- Demirseren, D.D.; Ceylan, G.; Akoglu, G.; Emre, S.; Erten, S.; Arman, A.; Metin, A. HLA-B51 subtypes in Turkish patients with Behcet’s disease and their correlation with clinical manifestations. Genet. Mol. Res. 2014, 13, 4788–4796. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, A.; Houman, M.H.; Massouadia, M.; Salem, T.B.; Khanfir, M.S.; Ghorbel, I.B.; Miled, M. Contribution of Hla-B51 in the susceptibility and specific clinical features of Behcet’s disease in Tunisian patients. Eur. J. Intern. Med. 2012, 23, 347–349. [Google Scholar] [CrossRef]
- Mizuki, N.; Yabuki, K.; Ota, M.; Verity, D.; Katsuyama, Y.; Ando, H.; Onari, K.; Goto, K.; Imagawa, Y.; Mandanat, W. Microsatellite mapping of a susceptible locus within the HLA region for Behcet’s disease using Jordanian patients. Hum. Immunol. 2001, 62, 186–190. [Google Scholar] [CrossRef]
- Castillo-Palma, M.J.; Sánchez Román, J.; Ocaña Medina, C.; González Escribano, M.F.; Núñez Roldán, A.; López-Checa, F. Serologic and molecular HLA typing in patients from Andalucia with Behcet’s disease. Genetic and clinical correlations. Med. Clin. 1996, 106, 121–125. [Google Scholar]
- Muñoz, S.A.; Orden, A.O.; Kostianovsky, A.; Pisoni, C.N.; Scolnik, M.; Luissi, A.; Bottinelli, Y.; Vijoditz, G.; Garcia, M.; Pena, C. The HLA-B*51 allele is strongly associated with Behcet disease in an argentinean population. Reumatol. Clín. 2020, 16, 282–285. [Google Scholar] [CrossRef]
- Rodríguez, M.; Walter, K.; Sanchez-Roman, J.; Garcfa-Lozano, J.; Núñez-Roldán, A. Association of HLA-B 51 subtypes and Behcet’s disease in Spain. Tissue Antigens 1998, 52, 78–80. [Google Scholar] [CrossRef]
- Sakly, N.; Boumiza, R.; Zrour-Hassen, S.; Hamzaoui, A.; Yahia, S.B.; Amara, H.; Khairallah, M.; Mahjoub, S.; Bergaoui, N.; Ghedira, I. HLA-B27 and HLA-B51 determination in Tunisian healthy subjects and patients with suspected ankylosing spondylitis and Behcet’s disease. Ann. N. Y. Acad. Sci. 2009, 1173, 564–569. [Google Scholar] [CrossRef]
- Sanz, L.; Gonzalez-Escribano, F.; De Pablo, R.; Nunez-Roldan, A.; Kreisler, M.; Vilches, C. HLA-Cw*1602: A new susceptibility marker of Behcet’s disease in southern Spain. Tissue Antigens 1998, 51, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Kirino, Y.; de Bakker, P.I.; Gül, A.; Kastner, D.L.; Remmers, E.F. Behçet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 8867–8872. [Google Scholar] [CrossRef] [PubMed]
- Capittini, C.; Rebuffi, C.; Lenti, M.V.; Di Sabatino, A.; Tinelli, C.; Martinetti, M.; De Silvestri, A. Global Meta-Analysis on the Association between Behcet Syndrome and Polymorphisms from the HLA Class I (A, B, and C) and Class II (DRB1, DQB1, and DPB1) Genes. Dis. Markers 2021, 2021, 9348697. [Google Scholar] [CrossRef]
- Kirino, Y.; Bertsias, G.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Sacli, F.S.; Erer, B.; Inoko, H.; et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat. Genet. 2013, 45, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Zhong, Z.; Zhou, Q.; Du, L.; Ye, Z.; Li, F.; Zhuang, W.; Wang, C.; Liang, L.; Ji, Y.; et al. Identification of Novel Risk Loci for Behçet’s Disease-Related Uveitis in a Chinese Population in a Genome-Wide Association Study. Arthritis Rheumatol. 2022, 74, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Al-Obeidi, A.F.; Nowatzky, J. Immunopathogenesis of Behcet’s disease. Clin. Immunol. 2023, 253, 109661. [Google Scholar] [CrossRef]
- Guasp, P.; Alvarez-Navarro, C.; Gomez-Molina, P.; Martín-Esteban, A.; Marcilla, M.; Barnea, E.; Admon, A.; de Castro, J.A. The Peptidome of Behçet’s Disease–Associated HLA–B*51:01 Includes Two Subpeptidomes Differentially Shaped by Endoplasmic Reticulum Aminopeptidase 1. Arthritis Rheumatol. 2016, 68, 505–515. [Google Scholar] [CrossRef]
- Kuiper, J.J.; Prinz, J.C.; Stratikos, E.; Kusnierczyk, P.; Arakawa, A.; Springer, S.; Mintoff, D.; Padjen, I.; Shumnalieva, R.; Vural, S.; et al. EULAR study group on ‘MHC-I-opathy’: Identifying disease-overarching mechanisms across disciplines and borders. Ann. Rheum. Dis. 2023, 82, 887–896. [Google Scholar] [CrossRef]
- McGonagle, D.; Aydin, S.Z.; Gül, A.; Mahr, A.; Direskeneli, H. ‘MHC-I-Opathy’-Unified Concept Spondyloarthritis Behçet Disease. Nat. Rev. Rheumatol. 2015, 11, 731–740. [Google Scholar] [CrossRef]
- Takeno, M. The association of Behcet’s syndrome with HLA-B51 as understood in 2021. Curr. Opin. Rheumatol. 2022, 34, 4–9. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.; Shahram, F.; Francisco, D.; Davatchi, F.; Abdollahi, B.S.; Ghaderibarmi, F.; Nadji, A.; Mojarad Shafiee, N.; Xavier, J.M.; Oliveira, S.A. Brief Report: Association of CCR1, KLRC4, IL12A–AS1, STAT4, and ERAP1 With Behçet’s Disease in Iranians. Arthritis Rheumatol. 2015, 67, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Paldino, G.; Paladini, F.; Mattorre, B.; Tuosto, L.; Sorrentino, R.; Fiorillo, M.T. The Impact of the ‘Mis-Peptidome’ on HLA Class I-Mediated Diseases: Contribution of ERAP1 and ERAP2 and Effects on the Immune Response. Int. J. Mol. Sci. 2020, 21, 9608. [Google Scholar] [CrossRef] [PubMed]
- López de Castro, J.A. How ERAP1 and ERAP2 Shape the Peptidomes of Disease-Associated MHC-I Proteins. Front. Immunol. 2018, 9, 2463. [Google Scholar] [CrossRef]
- Takeuchi, M.; Ombrello, M.J.; Kirino, Y.; Erer, B.; Tugal-Tutkun, I.; Seyahi, E.; Özyazgan, Y.; Watts, N.R.; Gül, A.; Kastner, D.L.; et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for Behçet’s disease in HLA-B*51 carriers. Ann. Rheum. Dis. 2016, 75, 2208–2211. [Google Scholar] [CrossRef]
- Cavers, A.; Kugler, M.C.; Ozguler, Y.; Al-Obeidi, A.F.; Hatemi, G.; Ueberheide, B.M.; Ucar, D.; Manches, O.; Nowatzky, J. Behcet’s disease risk-variant HLA-B51/ERAP1-Hap10 alters human CD8 T cell immunity. Ann. Rheum. Dis. 2022, 81, 1603–1611. [Google Scholar] [CrossRef]
- Guasp, P.; Barnea, E.; González-Escribano, M.F.; Jiménez-Reinoso, A.; Regueiro, J.R.; Admon, A.; López de Castro, J.A. The Behçet’s disease-associated variant of the aminopeptidase ERAP1 shapes a low-affinity HLA-B*51 peptidome by differential subpeptidome processing. J. Biol. Chem. 2017, 292, 9680–9689. [Google Scholar] [CrossRef]
- Cifaldi, L.; Romania, P.; Falco, M.; Lorenzi, S.; Meazza, R.; Petrini, S.; Andreani, M.; Pende, D.; Locatelli, F.; Fruci, D. ERAP1 regulates natural killer cell function by controlling the engagement of inhibitory receptors. Cancer Res. 2015, 75, 824–834. [Google Scholar] [CrossRef]
- Ryu, H.M.; Islam, S.M.S.; Sayeed, H.M.; Babita, R.; Seong, J.K.; Lee, H.; Sohn, S. Characterization of immune responses associated with ERAP-1 expression in HSV-induced Behcet’s disease mouse model. Clin. Immunol. 2023, 250, 109305. [Google Scholar] [CrossRef]
- Pepelyayeva, Y.; Rastall, D.P.W.; Aldhamen, Y.A.; O’Connell, P.; Raehtz, S.; Alyaqoub, F.S.; Blake, M.K.; Raedy, A.M.; Angarita, A.M.; Abbas, A.M.; et al. ERAP1 deficient mice have reduced Type 1 regulatory T cells and develop skeletal and intestinal features of Ankylosing Spondylitis. Sci. Rep. 2018, 8, 12464. [Google Scholar] [CrossRef]
- Gebreselassie, D.; Spiegel, H.; Vukmanovic, S. Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum. Immunol. 2006, 67, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Falk, K.; Rötzschke, O.; Takiguchi, M.; Gnau, V.; Stevanović, S.; Jung, G.; Rammensee, H.-G. Peptide motifs of HLA-B51,-B52 and-B78 molecules, and implications for Behcet’s disease. Int. Immunol. 1995, 7, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Gur, M.; Golcuk, M.; Gul, A.; Erman, B. Molecular dynamics simulations provide molecular insights into the role of HLA-B51 in Behçet’s disease pathogenesis. Chem. Biol. Drug Des. 2020, 96, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Huyton, T.; Ladas, N.; Schumacher, H.; Blasczyk, R.; Bade-Doeding, C. Pocketcheck: Updating the HLA class I peptide specificity roadmap. Tissue Antigens 2012, 80, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Sanjanwala, B.; Draghi, M.; Norman, P.J.; Guethlein, L.A.; Parham, P. Polymorphic sites away from the Bw4 epitope that affect interaction of Bw4+ HLA-B with KIR3DL1. J. Immunol. 2008, 181, 6293–6300. [Google Scholar] [CrossRef]
- Kang, E.H.; Kim, J.Y.; Takeuchi, F.; Kim, J.W.; Shin, K.; Lee, E.Y.; Lee, Y.J.; Lee, E.B.; Park, M.H.; Song, Y.W. Associations between the HLA-A polymorphism and the clinical manifestations of Behcet’s disease. Arthritis Res. Ther. 2011, 13, R49. [Google Scholar] [CrossRef]
- Kuranov, A.B.; Kotter, I.; Henes, J.C.; Abisheva, S.T.; Steiert, I.; Riewerts, F.; Momynaliev, K.T.; Muller, C.A. Behcet’s disease in HLA-B*51 negative Germans and Turks shows association with HLA-Bw4-80I. Arthritis Res. Ther. 2014, 16, R116. [Google Scholar] [CrossRef]
- Petrushkin, H.; Norman, P.J.; Lougee, E.; Parham, P.; Wallace, G.R.; Stanford, M.R.; Fortune, F. KIR3DL1/S1 Allotypes Contribute Differentially to the Development of Behcet Disease. J. Immunol. 2019, 203, 1629–1635. [Google Scholar] [CrossRef]
- Single, R.M.; Martin, M.P.; Gao, X.; Meyer, D.; Yeager, M.; Kidd, J.R.; Kidd, K.K.; Carrington, M. Global diversity and evidence for coevolution of KIR and HLA. Nat. Genet. 2007, 39, 1114–1119. [Google Scholar] [CrossRef]
- Erer, B.; Takeuchi, M.; Ustek, D.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Duymaz-Tozkir, J.; Gul, A.; Kastner, D.L.; Remmers, E.F.; et al. Evaluation of KIR3DL1/KIR3DS1 polymorphism in Behcet’s disease. Genes. Immun. 2016, 17, 396–399. [Google Scholar] [CrossRef]
- Mizuki, N.; Meguro, A.; Ota, M.; Ohno, S.; Shiota, T.; Kawagoe, T.; Ito, N.; Kera, J.; Okada, E.; Yatsu, K.; et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet’s disease susceptibility loci. Nat. Genet. 2010, 42, 703–706. [Google Scholar] [CrossRef]
- Touitou, I.; Magne, X.; Molinari, N.; Navarro, A.; Quellec, A.L.; Picco, P.; Seri, M.; Ozen, S.; Bakkaloglu, A.; Karaduman, A.; et al. MEFV mutations in Behcet’s disease. Hum. Mutat. 2000, 16, 271–272. [Google Scholar] [CrossRef]
- Atagunduz, P.; Ergun, T.; Direskeneli, H. MEFV mutations are increased in Behcet’s disease (BD) and are associated with vascular involvement. Clin. Exp. Rheumatol. 2003, 21, S35–S37. [Google Scholar]
- Kirino, Y.; Zhou, Q.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Özyazgan, Y.; Ugurlu, S.; Erer, B.; Abaci, N.; et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behçet disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8134–8139. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Gundogdu, M.; Gokpinar Ili, E.; Durmaz, C.D.; Vural, A.; Steinmuller-Magin, L.; Kleinhempel, A.; Holdt, L.M.; Ruzicka, T.; Giehl, K.A.; et al. Association of pyrin mutations and autoinflammation with complex phenotype hidradenitis suppurativa: A case-control study. Br. J. Dermatol. 2019, 180, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Shindo, A.; Ii, Y.; Kishida, D.; Niwa, A.; Nishiguchi, Y.; Matsuura, K.; Kato, N.; Mizutani, A.; Tachibana, K.; et al. MEFV gene mutations in neuro-Behcet’s disease and neuro-Sweet disease. Ann. Clin. Transl. Neurol. 2019, 6, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Cho, Y.H.; Lee, G.S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liao, D.; Yang, L.; Hou, S. Association between Functional MICA-TM and Behcet’s Disease: A Systematic Review and Meta-analysis. Sci. Rep. 2016, 6, 21033. [Google Scholar] [CrossRef]
- Xavier, J.M.; Shahram, F.; Sousa, I.; Davatchi, F.; Matos, M.; Abdollahi, B.S.; Sobral, J.; Nadji, A.; Oliveira, M.; Ghaderibarim, F.; et al. FUT2: Filling the gap between genes and environment in Behçet’s disease? Ann. Rheum. Dis. 2015, 74, 618–624. [Google Scholar] [CrossRef]
- Mehmood, N.; Low, L.; Wallace, G.R. Behçet’s Disease—Do Microbiomes and Genetics Collaborate in Pathogenesis? Front. Immunol. 2021, 12, 648341. [Google Scholar] [CrossRef]
- Ferrer-Admetlla, A.; Sikora, M.; Laayouni, H.; Esteve, A.; Roubinet, F.; Blancher, A.; Calafell, F.; Bertranpetit, J.; Casals, F. A Natural History of FUT2 Polymorphism in Humans. Mol. Biol. Evol. 2009, 26, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Franzosa, E.A.; Rahnavard, G.; Hall, A.B.; Vlamakis, H.; Stevens, C.; Daly, M.J.; Xavier, R.J.; Huttenhower, C. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Meguro, A.; Ota, M.; Kitaichi, N.; Katsuyama, Y.; Takemoto, Y.; Namba, K.; Yoshida, K.; Song, Y.W.; Park, K.S.; et al. Association of TLR4 polymorphisms with Behcet’s disease in a Korean population. Rheumatology 2009, 48, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Meguro, A.; Ota, M.; Katsuyama, Y.; Oka, A.; Ohno, S.; Inoko, H.; Mizuki, N. Association of the toll-like receptor 4 gene polymorphisms with Behçet’s disease. Ann. Rheum. Dis. 2008, 67, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Hu, R.; Hou, S.; Ye, Z.; Xiang, Q.; Qi, J.; Zhou, Y.; Kijlstra, A.; Yang, P. Association of TLR2 gene polymorphisms with ocular Behcet’s disease in a Chinese Han population. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8384–8392. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, N.; Kirino, Y.; Soejima, Y.; Onodera, M.; Arai, K.; Tamura, E.; Ishikawa, T.; Kawai, T.; Uchiyama, T.; Nomura, S.; et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease. Arthritis Res. Ther. 2019, 21, 137. [Google Scholar] [CrossRef]
- Li, H.; Liu, Q.; Hou, S.; Du, L.; Zhou, Q.; Zhou, Y.; Kijlstra, A.; Li, Z.; Yang, P. TNFAIP3 gene polymorphisms confer risk for Behcet’s disease in a Chinese Han population. Hum. Genet. 2013, 132, 293–300. [Google Scholar] [CrossRef]
- Deng, Y.; Zhu, W.; Zhou, X. Immune Regulatory Genes Are Major Genetic Factors to Behcet Disease: Systematic Review. Open Rheumatol. J. 2018, 12, 70–85. [Google Scholar] [CrossRef]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef]
- Hitotsumatsu, O.; Ahmad, R.C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008, 28, 381–390. [Google Scholar] [CrossRef]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Cotsapas, C.; Davies, L.; Price, A.L.; de Bakker, P.I.; Maller, J.; Pe’er, I.; Burtt, N.P.; Blumenstiel, B.; DeFelice, M.; et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat. Genet. 2007, 39, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Deng, M.; Gan, C.; Wang, L.; Mao, H.; Li, Q. A Novel Missense Mutation in TNFAIP3 causes Haploinsufficiency of A20. Cell. Immunol. 2021, 371, 104453. [Google Scholar] [CrossRef]
- Berteau, F.; Rouviere, B.; Delluc, A.; Nau, A.; Le Berre, R.; Sarrabay, G.; Touitou, I.; de Moreuil, C. Autosomic dominant familial Behçet disease and haploinsufficiency A20: A review of the literature. Autoimmun. Rev. 2018, 17, 809–815. [Google Scholar] [CrossRef]
- Korman, B.D.; Kastner, D.L.; Gregersen, P.K.; Remmers, E.F. STAT4: Genetics, mechanisms, and implications for autoimmunity. Curr. Allergy Asthma Rep. 2008, 8, 398–403. [Google Scholar] [CrossRef]
- Hou, S.; Yang, Z.; Du, L.; Jiang, Z.; Shu, Q.; Chen, Y.; Li, F.; Zhou, Q.; Ohno, S.; Chen, R.; et al. Identification of a susceptibility locus in STAT4 for Behçet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012, 64, 4104–4113. [Google Scholar] [CrossRef]
- Esin, S.; Gul, A.; Hodara, V.; Jeddi-Tehrani, M.; Dilsen, N.; Konice, M.; Andersson, R.; Wigzell, H. Peripheral blood T cell expansions in patients with Behcet’s disease. Clin. Exp. Immunol. 1997, 107, 520–527. [Google Scholar] [CrossRef]
- Direskeneli, H.; Eksioglu-Demiralp, E.; Kibaroglu, A.; Yavuz, S.; Ergun, T.; Akoglu, T. Oligoclonal T cell expansions in patients with Behcet’s disease. Clin. Exp. Immunol. 1999, 117, 166–170. [Google Scholar] [CrossRef]
- Lehner, T.; Lavery, E.; Smith, R.; van der Zee, R.; Mizushima, Y.; Shinnick, T. Association between the 65-kilodalton heat shock protein, Streptococcus sanguis, and the corresponding antibodies in Behçet’s syndrome. Infect. Immun. 1991, 59, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Kitteringham, N.; Shiba, K.; Goseki, M.; Kimura, A.; Mineshita, S. Use of the highly sensitive PCR method to detect the Herpes simplex virus type 1 genome and its expression in samples from Behçet disease patients. J. Med. Dent. Sci. 1998, 45, 51–58. [Google Scholar] [PubMed]
- Kaneko, F.; Oyama, N.; Yanagihori, H.; Isogai, E.; Yokota, K.; Oguma, K. The role of streptococcal hypersensitivity in the pathogenesis of Behçet’s Disease. Eur. J. Dermatol. EJD 2008, 18, 489–498. [Google Scholar] [CrossRef]
- Cheng, L.; Zhan, H.; Liu, Y.; Chen, H.; Zhang, F.; Zheng, W.; Li, Y. Infectious agents and pathogenesis of Behcet’s disease: An extensive review. Clin. Immunol. 2023, 251, 109631. [Google Scholar] [CrossRef] [PubMed]
- Direskeneli, H.; Eksioglu-Demiralp, E.; Yavuz, S.; Ergun, T.; Shinnick, T.; Lehner, T.; Akoglu, T. T cell responses to 60/65 kDa heat shock protein derived peptides in Turkish patients with Behcet’s disease. J. Rheumatol. 2000, 27, 708–713. [Google Scholar] [PubMed]
- de Chambrun, M.P.; Wechsler, B.; Geri, G.; Cacoub, P.; Saadoun, D. New insights into the pathogenesis of Behcet’s disease. Autoimmun. Rev. 2012, 11, 687–698. [Google Scholar] [CrossRef]
- Yazici, H.; Seyahi, E.; Hatemi, G.; Yazici, Y. Behçet syndrome: A contemporary view. Nat. Rev. Rheumatol. 2018, 14, 107–119. [Google Scholar] [CrossRef]
- Lule, S.; Colpak, A.I.; Balci-Peynircioglu, B.; Gursoy-Ozdemir, Y.; Peker, S.; Kalyoncu, U.; Can, A.; Tekin, N.; Demiralp, D.; Dalkara, T. Behçet disease serum is immunoreactive to neurofilament medium which share common epitopes to bacterial HSP-65, a putative trigger. J. Autoimmun. 2017, 84, 87–96. [Google Scholar] [CrossRef]
- Kibaroglu, A.; Eksioglu-Demiralp, E.; Akoglu, T.; Direskeneli, H. T and NK cell subset changes with microbial extracts and human HSP60-derived peptides in Behçet’s disease. Clin. Exp. Rheumatol. 2004, 22, S59–S63. [Google Scholar]
- Pervin, K.; Childerstone, A.; Shinnick, T.; Mizushima, Y.; van der Zee, R.; Hasan, A.; Vaughan, R.; Lehner, T. T cell epitope expression of mycobacterial and homologous human 65-kilodalton heat shock protein peptides in short term cell lines from patients with Behçet’s disease. J. Immunol. 1993, 151, 2273–2282. [Google Scholar] [CrossRef]
- Hasan, A.; Fortune, F.; Wilson, A.; Warr, K.; Lehner, T.; Sanderson, J.; Shinnick, T.; Mizushima, Y.; van der Zee, R.; Stanford, M. Role of γδ T cells in pathogenesis and diagnosis of Behçet’s disease. Lancet 1996, 347, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Yasuoka, H.; Okazaki, Y.; Kawakami, Y.; Hirakata, M.; Inoko, H.; Ikeda, Y.; Kuwana, M. Autoreactive CD8+ cytotoxic T lymphocytes to major histocompatibility complex class I chain–related gene A in patients with Behçet’s disease. Arthritis Rheum. 2004, 50, 3658–3662. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, A.; Emmi, G.; Low, L.; Sofi, F.; Wallace, G.R. Microbiome in Behcet’s syndrome. Clin. Immunol. 2023, 250, 109304. [Google Scholar] [CrossRef] [PubMed]
- Ogunkolade, W.; Senusi, A.A.; Desai, P.; Sacoor, S.; Bibi, A.; Gokani, B.; Sandionigi, A.; Fortune, F. Profiling the microbiome of oral and genital mucosal surfaces in Behcet’s disease. Clin. Immunol. 2023, 253, 109654. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Kerl, K.; Ertop Dogan, P.; Vollmer, S.; Puchta, U.; He, M.; Arakawa, Y.; Heper, A.O.; Karal-Oktem, A.; Hartmann, D.; et al. Lesional activation of T(c) 17 cells in Behcet disease and psoriasis supports HLA class I-mediated autoimmune responses. Br. J. Dermatol. 2021, 185, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takahashi, H.; Satoh, T.; Okazaki, Y.; Mizuki, N.; Takahashi, K.; Ikezawa, Z.; Kuwana, M. Natural killer cells control a T-helper 1 response in patients with Behçet’s disease. Arthritis Res. Ther. 2010, 12, R80. [Google Scholar] [CrossRef]
- Yasuoka, H.; Yamaguchi, Y.; Mizuki, N.; Nishida, T.; Kawakami, Y.; Kuwana, M. Preferential activation of circulating CD8+ and gammadelta T cells in patients with active Behçet’s disease and HLA-B51. Clin. Exp. Rheumatol. 2008, 26, S59–S63. [Google Scholar]
- Yu, H.G.; Lee, D.S.; Seo, J.M.; Ahn, J.K.; Yu, Y.S.; Lee, W.J.; Chung, H. The number of CD8+ T cells and NKT cells increases in the aqueous humor of patients with Behçet’s uveitis. Clin. Exp. Immunol. 2004, 137, 437–443. [Google Scholar] [CrossRef]
- Takase, H.; Sugita, S.; Taguchi, C.; Imai, Y.; Mochizuki, M. Capacity of ocular infiltrating T helper type 1 cells of patients with non-infectious uveitis to produce chemokines. Br. J. Ophthalmol. 2006, 90, 765–768. [Google Scholar] [CrossRef]
- Ye, Z.; Deng, B.; Wang, C.; Zhang, D.; Kijlstra, A.; Yang, P. Decreased B and T lymphocyte attenuator in Behcet’s disease may trigger abnormal Th17 and Th1 immune responses. Sci. Rep. 2016, 6, 20401. [Google Scholar] [CrossRef]
- Tong, B.; Liu, X.; Xiao, J.; Su, G. Immunopathogenesis of Behcet’s Disease. Front. Immunol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: Regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Lucherini, O.M.; Lopalco, G.; Cantarini, L.; Emmi, G.; Lopalco, A.; Venerito, V.; Vitale, A.; Iannone, F. Critical regulation of Th17 cell differentiation by serum amyloid-A signalling in Behcet’s disease. Immunol. Lett. 2018, 201, 38–44. [Google Scholar] [CrossRef]
- Shimizu, J.; Takai, K.; Takada, E.; Fujiwara, N.; Arimitsu, N.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; Suzuki, N. Possible association of proinflammatory cytokines including IL1β and TNFα with enhanced Th17 cell differentiation in patients with Behcet’s disease. Clin. Rheumatol. 2016, 35, 1857–1863. [Google Scholar] [CrossRef]
- Ahmadi, M.; Yousefi, M.; Abbaspour-Aghdam, S.; Dolati, S.; Aghebati-Maleki, L.; Eghbal-Fard, S.; Khabbazi, A.; Rostamzadeh, D.; Alipour, S.; Shabani, M.; et al. Disturbed Th17/Treg balance, cytokines, and miRNAs in peripheral blood of patients with Behcet’s disease. J. Cell. Physiol. 2019, 234, 3985–3994. [Google Scholar] [CrossRef]
- Berzins, S.P.; Ritchie, D.S. Natural killer T cells: Drivers or passengers in preventing human disease? Nat. Rev. Immunol. 2014, 14, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The Biology of NKT Cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Cameron, G.; Pellicci, D.G.; Uldrich, A.P.; Besra, G.S.; Illarionov, P.; Williams, S.J.; La Gruta, N.L.; Rossjohn, J.; Godfrey, D.I. Antigen Specificity of Type I NKT Cells Is Governed by TCR β-Chain Diversity. J. Immunol. 2015, 195, 4604–4614. [Google Scholar] [CrossRef] [PubMed]
- Bonacini, M.; Soriano, A.; Zerbini, A.; Calò, E.; Cimino, L.; Muratore, F.; Fontana, L.; Braglia, L.; Parmeggiani, M.; Salvarani, C.; et al. Higher Frequencies of Lymphocytes Expressing the Natural Killer Group 2D Receptor in Patients with Behçet Disease. Front. Immunol. 2018, 9, 2157. [Google Scholar] [CrossRef]
- Hamzaoui, K.; Kamoun, M.; Houman, H.; Hentati, F.; Hamza, M.H.; Ayed, K.; Hamzaoui, A. Discrepancies of NKT cells expression in peripheral blood and in cerebrospinal fluid from Behçet’s disease. J. Neuroimmunol. 2006, 175, 160–168. [Google Scholar] [CrossRef]
- Kalyan, S.; Kabelitz, D. Defining the nature of human γδ T cells: A biographical sketch of the highly empathetic. Cell. Mol. Immunol. 2013, 10, 21–29. [Google Scholar] [CrossRef]
- Chien, Y.-h.; Meyer, C.; Bonneville, M. γδ T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Parlakgul, G.; Guney, E.; Erer, B.; Kılıcaslan, Z.; Direskeneli, H.; Gul, A.; Saruhan-Direskeneli, G. Expression of regulatory receptors on γδ T Cells and their cytokine production in Behcet’s disease. Arthritis Res. Ther. 2013, 15, R15. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Bioley, G.; Lévy, N.; Eberl, M.; Luo, M.; Tampé, R.; Lévy, F.; Romero, P.; Moser, B. Cross-presenting human γδ T cells induce robust CD8+ αβ T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Clemente, A.; Cambra, A.; Munoz-Saá, I.; Crespí, C.; Pallarés, L.; Juan, A.; Matamoros, N.; Julià, M.R. Phenotype markers and cytokine intracellular production by CD8+ gammadelta T lymphocytes do not support a regulatory T profile in Behçet’s disease patients and healthy controls. Immunol. Lett. 2010, 129, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Dieli, F.; Caccamo, N. Immunomodulatory role of statins in autoimmune disease: Is there a role for human γδT cells? Nat. Rev. Immunol. 2006, 6, 564. [Google Scholar] [CrossRef]
- Accardo-Palumbo, A.; Ferrante, A.; Cadelo, M.; Ciccia, F.; Parrinello, G.; Lipari, L.; Giardina, A.R.; Riili, M.; Giardina, E.; Dieli, F.; et al. The level of soluble Granzyme A is elevated in the plasma and in the Vgamma9/Vdelta2 T cell culture supernatants of patients with active Behçet’s disease. Clin. Exp. Rheumatol. 2004, 22, S45–S49. [Google Scholar] [PubMed]
- Fortune, F.; Walker, J.; Lehner, T. The expression of gamma delta T cell receptor and the prevalence of primed, activated and IgA-bound T cells in Behçet’s syndrome. Clin. Exp. Immunol. 1990, 82, 326–332. [Google Scholar] [CrossRef]
- Suzuki, Y.; Hoshi, K.; Matsuda, T.; Mizushima, Y. Increased peripheral blood gamma delta+ T cells and natural killer cells in Behçet’s disease. J. Rheumatol. 1992, 19, 588–592. [Google Scholar]
- Freysdottir, J.; Lau, S.; Fortune, F. Gammadelta T cells in Behçet’s disease (BD) and recurrent aphthous stomatitis (RAS). Clin. Exp. Immunol. 1999, 118, 451–457. [Google Scholar] [CrossRef]
- Bank, I.; Duvdevani, M.; Livneh, A. Expansion of gammadelta T-cells in Behçet’s disease: Role of disease activity and microbial flora in oral ulcers. J. Lab. Clin. Med. 2003, 141, 33–40. [Google Scholar] [CrossRef]
- Ergun, T.; İnce, Ü.; Ekşioğlu-Demiralp, E.; Direskeneli, H.; Gürbüz, O.; Gürses, L.; Aker, F.; Akoğlu, T. HSP 60 expression in mucocutaneous lesions of Behçet’s disease. J. Am. Acad. Dermatol. 2001, 45, 904–909. [Google Scholar] [CrossRef]
- Triolo, G.; Accardo-Palumbo, A.; Dieli, F.; Ciccia, F.; Ferrante, A.; Giardina, E.; Sano, C.D.; Licata, G. Vgamma9/Vdelta2 T lymphocytes in Italian patients with Behçet’s disease: Evidence for expansion, and tumour necrosis factor receptor II and interleukin-12 receptor beta1 expression in active disease. Arthritis Res. Ther. 2003, 5, R262–R268. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Hill, J.A.; Mathis, D.; Benoist, C. Foxp3+ regulatory T cells: Differentiation, specification, subphenotypes. Nat. Immunol. 2009, 10, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Geri, G.; Terrier, B.; Rosenzwajg, M.; Wechsler, B.; Touzot, M.; Seilhean, D.; Tran, T.-A.; Bodaghi, B.; Musset, L.; Soumelis, V.; et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behçet disease. J. Allergy Clin. Immunol. 2011, 128, 655–664. [Google Scholar] [CrossRef]
- Hamzaoui, K.; Borhani Haghighi, A.; Ghorbel, I.B.; Houman, H. RORC and Foxp3 axis in cerebrospinal fluid of patients with Neuro-Behçet’s Disease. J. Neuroimmunol. 2011, 233, 249–253. [Google Scholar] [CrossRef]
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar] [CrossRef]
- Tian, Z.; Gershwin, M.E.; Zhang, C. Regulatory NK cells in autoimmune disease. J. Autoimmun. 2012, 39, 206–215. [Google Scholar] [CrossRef]
- French, A.R.; Yokoyama, W.M. Natural killer cells and autoimmunity. Arthritis Res. Ther. 2003, 6, 8. [Google Scholar] [CrossRef]
- Hasan, M.S.; Ryan, P.L.; Bergmeier, L.A.; Fortune, F. Circulating NK cells and their subsets in Behçet’s disease. Clin. Exp. Immunol. 2017, 188, 311–322. [Google Scholar] [CrossRef]
- Cosan, F.; Aktas Cetin, E.; Akdeniz, N.; Emrence, Z.; Cefle, A.; Deniz, G. Natural Killer Cell Subsets and Their Functional Activity in Behçet’s Disease. Immunol. Investig. 2017, 46, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Kucuksezer, U.C.; Aktas-Cetin, E.; Bilgic-Gazioglu, S.; Tugal-Tutkun, I.; Gul, A.; Deniz, G. Natural killer cells dominate a Th-1 polarized response in Behçet’s disease patients with uveitis. Clin. Exp. Rheumatol. 2015, 33, S24–S29. [Google Scholar]
- Kawakami, T.; Ohashi, S.; Kawa, Y.; Takahama, H.; Ito, M.; Soma, Y.; Mizoguchi, M. Elevated Serum Granulocyte Colony-Stimulating Factor Levels in Patients With Active Phase of Sweet Syndrome and Patients With Active Behçet Disease: Implication in Neutrophil Apoptosis Dysfunction. Arch. Dermatol. 2004, 140, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Balta, S.; Ozturk, C.; Balta, I.; Demirkol, S.; Demir, M.; Celik, T.; Iyisoy, A. The neutrophil–lymphocyte ratio and inflammation. Angiology 2016, 67, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Djaballah-Ider, F.; Touil-Boukoffa, C. Effect of combined colchicine-corticosteroid treatment on neutrophil/lymphocyte ratio: A predictive marker in Behçet disease activity. Inflammopharmacology 2020, 28, 819–829. [Google Scholar] [CrossRef]
- Le Joncour, A.; Martos, R.; Loyau, S.; Lelay, N.; Dossier, A.; Cazes, A.; Fouret, P.; Domont, F.; Papo, T.; Jandrot-Perrus, M.; et al. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann. Rheum. Dis. 2019, 78, 1274–1282. [Google Scholar] [CrossRef]
- Mendoza-Pinto, C.; García-Carrasco, M.; Jiménez-Hernández, M.; Jiménez Hernández, C.; Riebeling-Navarro, C.; Nava Zavala, A.; Vera Recabarren, M.; Espinosa, G.; Jara Quezada, J.; Cervera, R. Etiopathogenesis of Behcet’s disease. Autoimmun. Rev. 2010, 9, 241–245. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoshbakht, S.; Başkurt, D.; Vural, A.; Vural, S. Behçet’s Disease: A Comprehensive Review on the Role of HLA-B*51, Antigen Presentation, and Inflammatory Cascade. Int. J. Mol. Sci. 2023, 24, 16382. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242216382
Khoshbakht S, Başkurt D, Vural A, Vural S. Behçet’s Disease: A Comprehensive Review on the Role of HLA-B*51, Antigen Presentation, and Inflammatory Cascade. International Journal of Molecular Sciences. 2023; 24(22):16382. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242216382
Chicago/Turabian StyleKhoshbakht, Saba, Defne Başkurt, Atay Vural, and Seçil Vural. 2023. "Behçet’s Disease: A Comprehensive Review on the Role of HLA-B*51, Antigen Presentation, and Inflammatory Cascade" International Journal of Molecular Sciences 24, no. 22: 16382. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242216382