
Conjugates of Tacrine and Salicylic Acid Derivatives as New Promising Multitarget Agents for Alzheimer’s Disease

 , , , , , , , , and
, , , , , , , , and 
Abstract
:1. Introduction
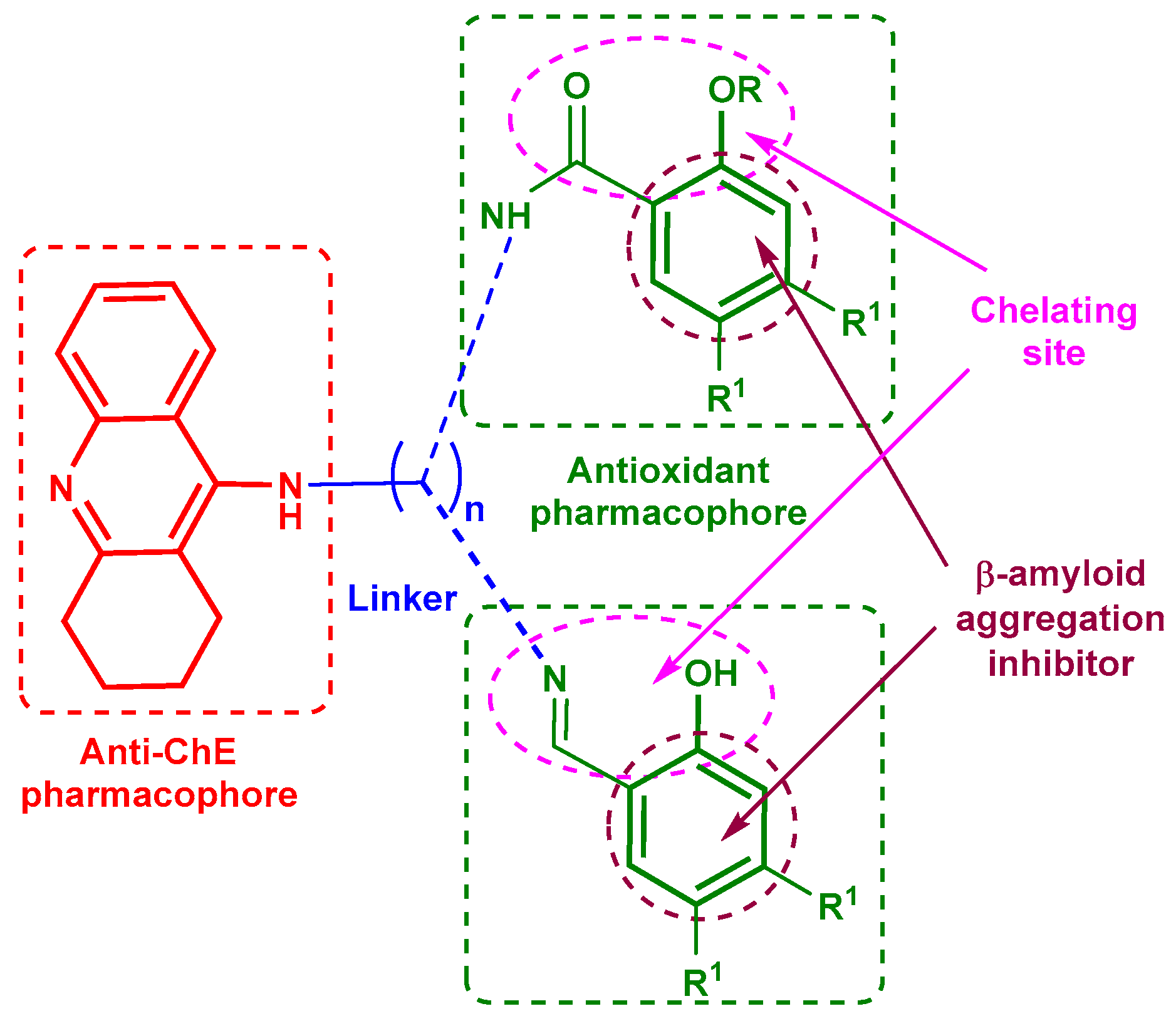
2. Results and Discussion
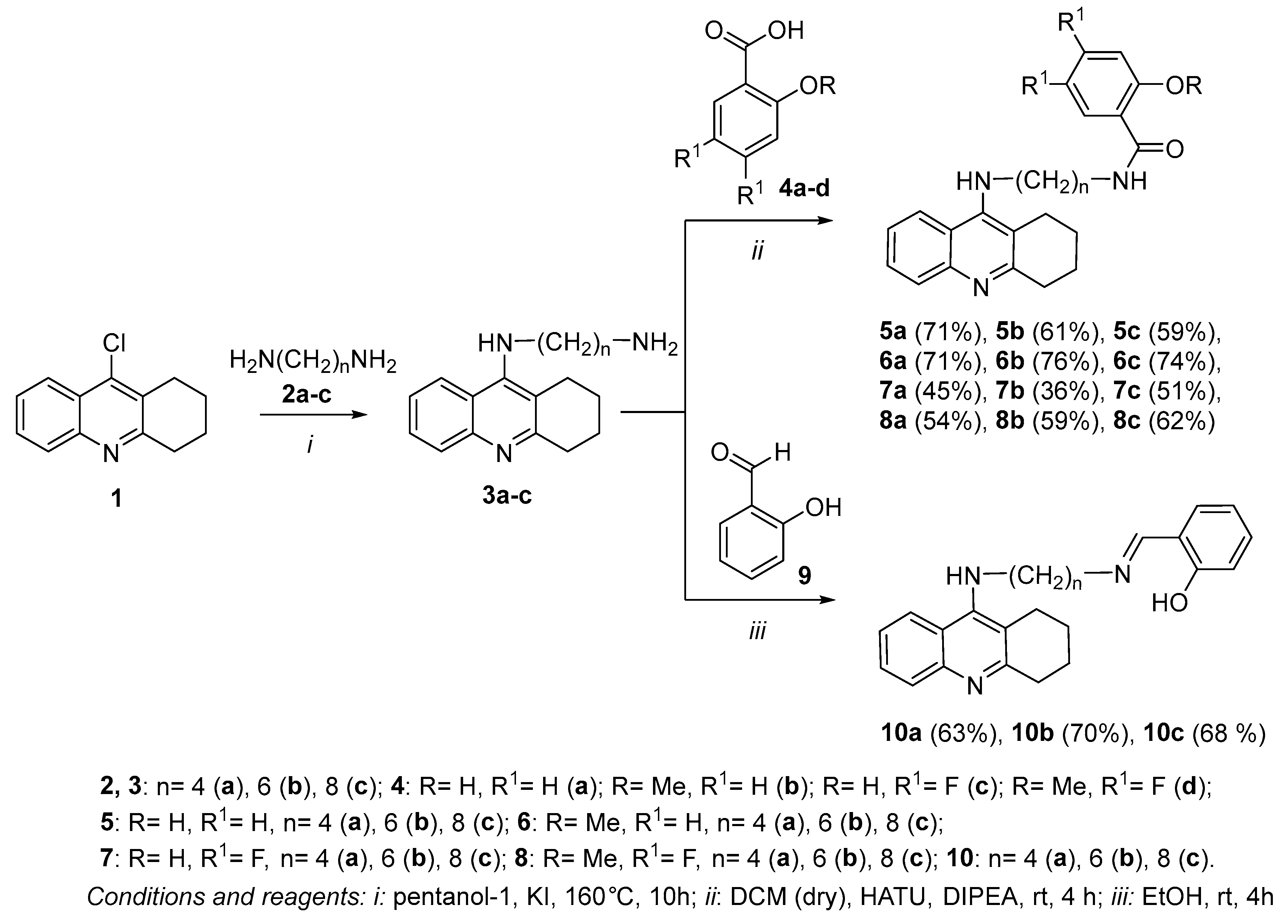
2.1. Chemistry
2.2. Biological Investigations
2.2.1. Inhibition Studies of AChE, BChE, and CES. Structure–Activity Relationships
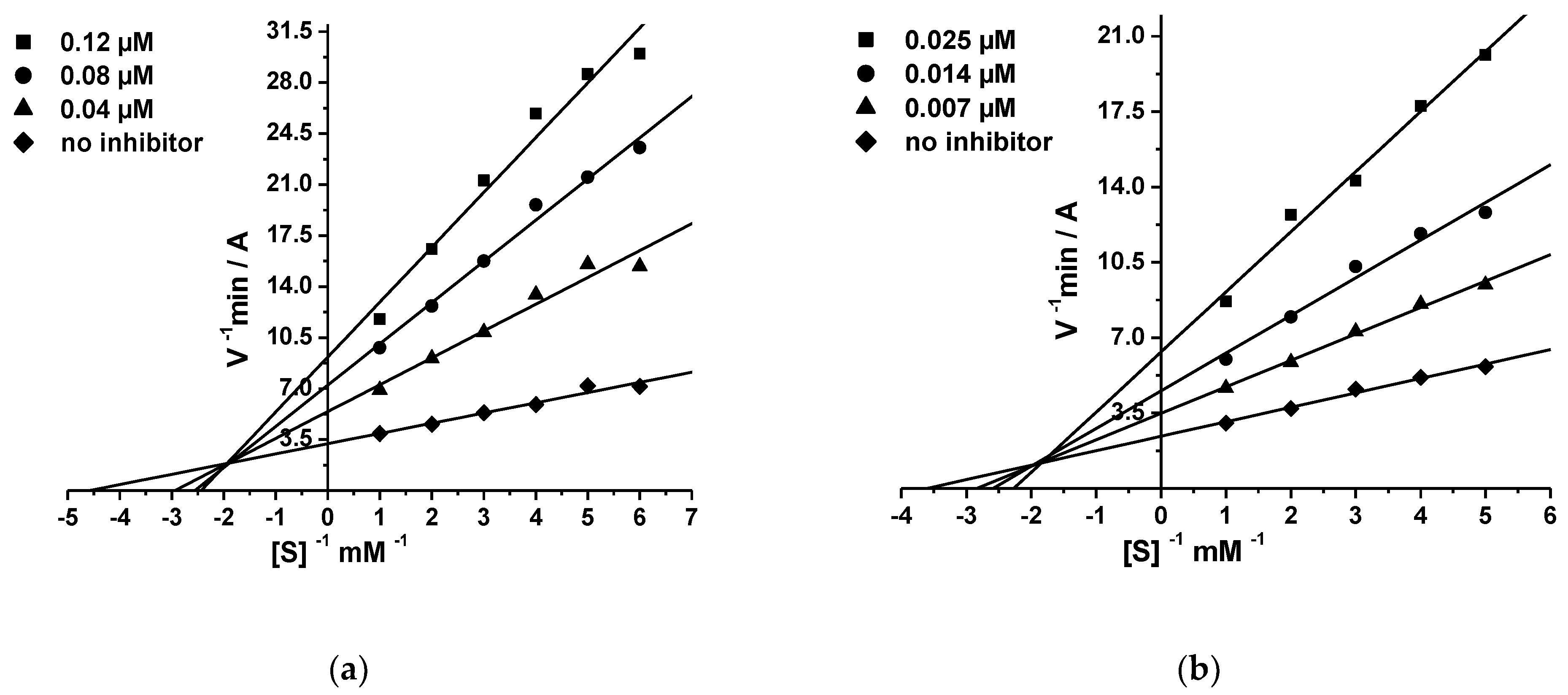
2.2.2. Kinetic Studies of AChE and BChE Inhibition
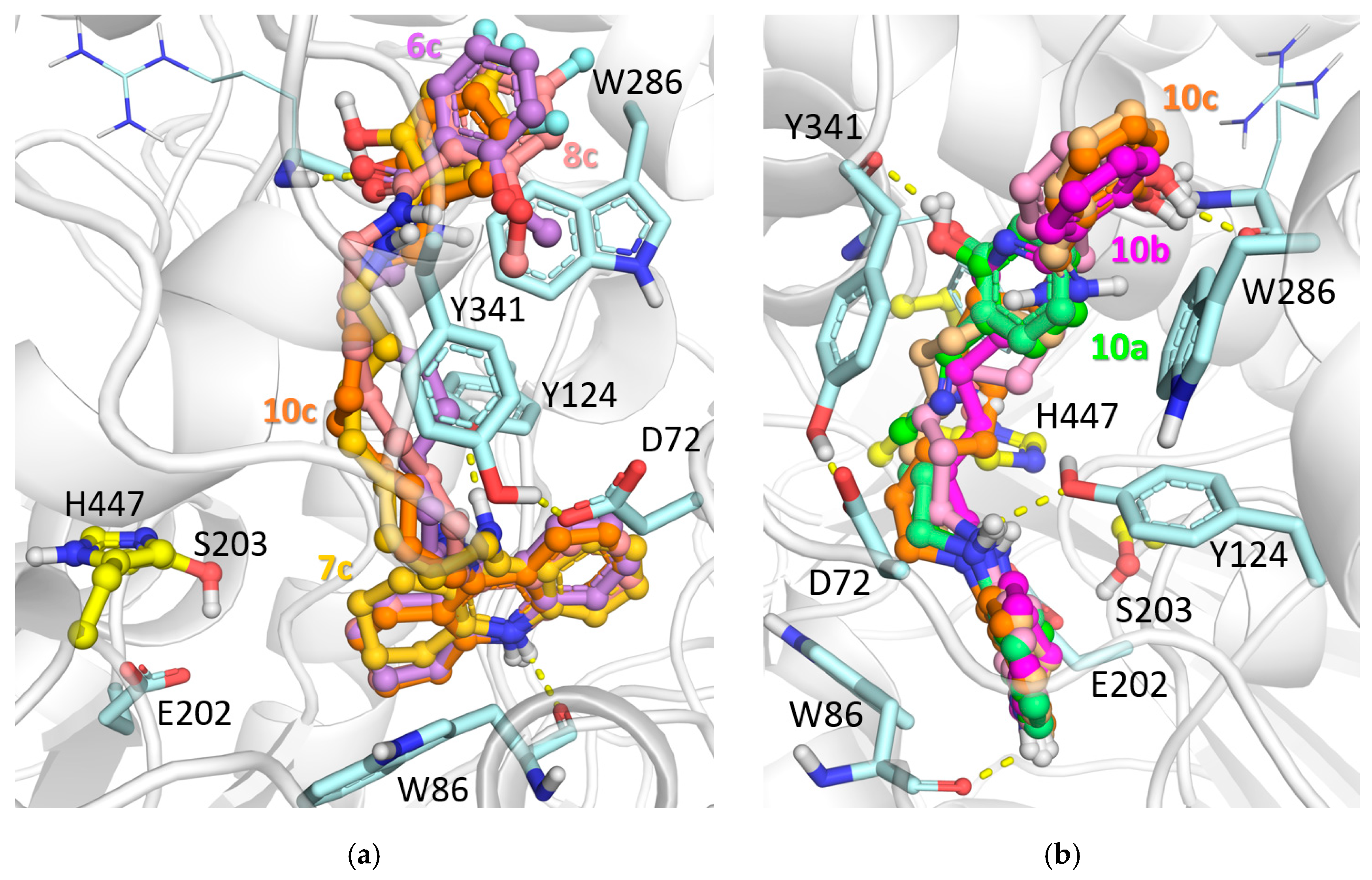
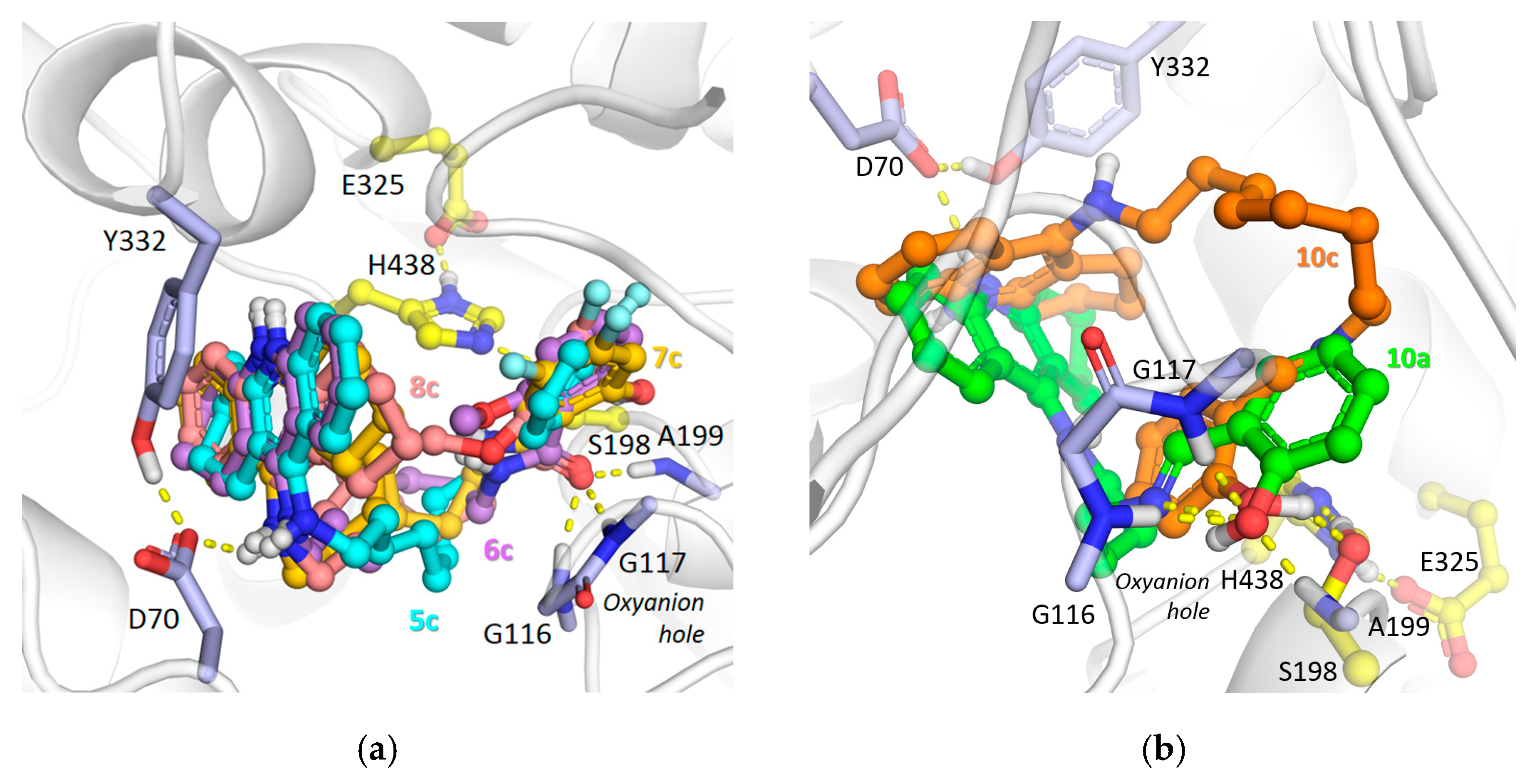
2.2.3. Molecular Modeling Studies
2.2.4. Displacement of Propidium Iodide from the PAS of EeAChE
2.2.5. Inhibition of β-Amyloid (1–42) (Aβ42) Self-Aggregation
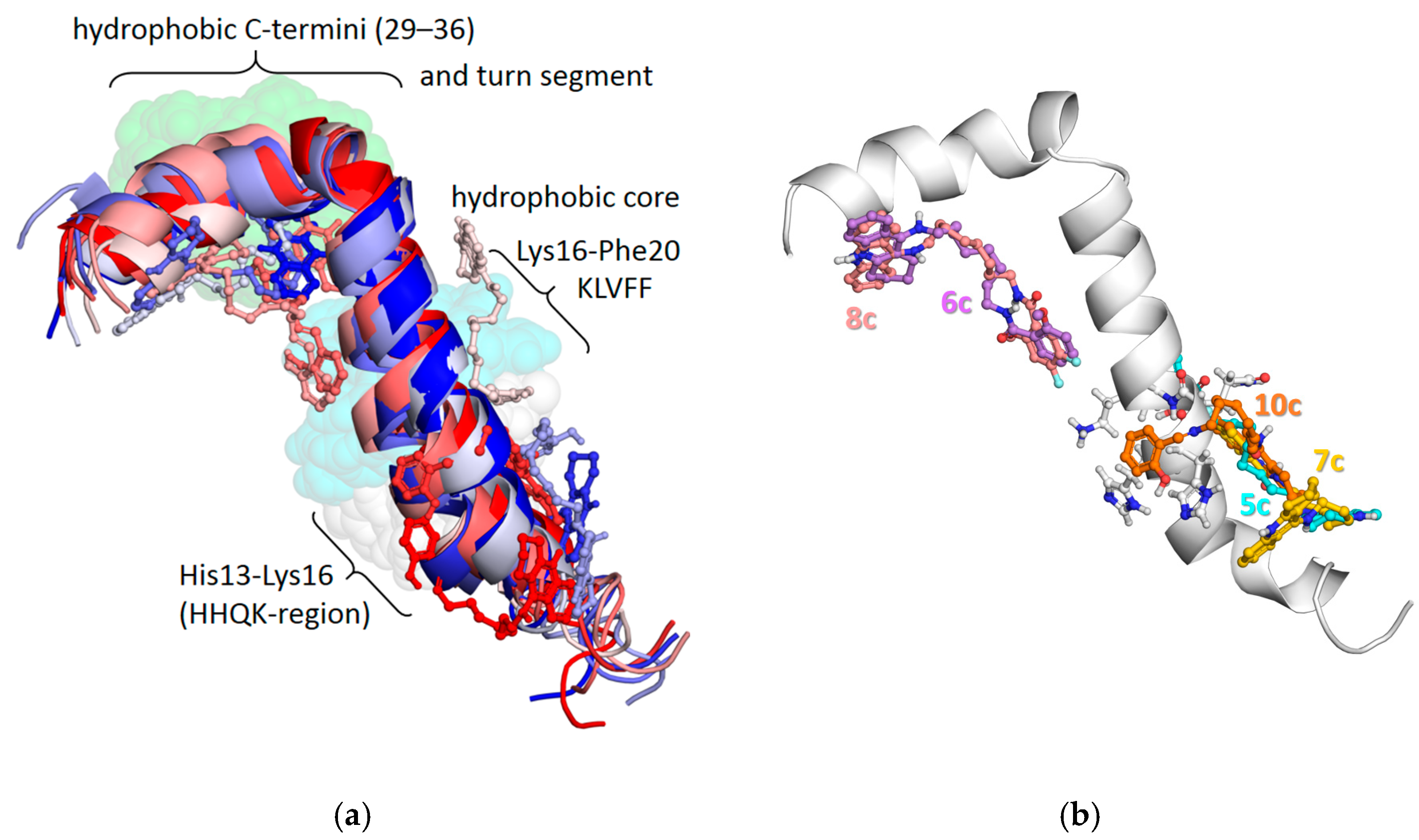
2.2.6. Molecular Modeling: Interactions of Conjugates with Aβ42
2.2.7. Antioxidant (AO) Activity
2.2.8. Quantum Chemical Analyses of AO Activity
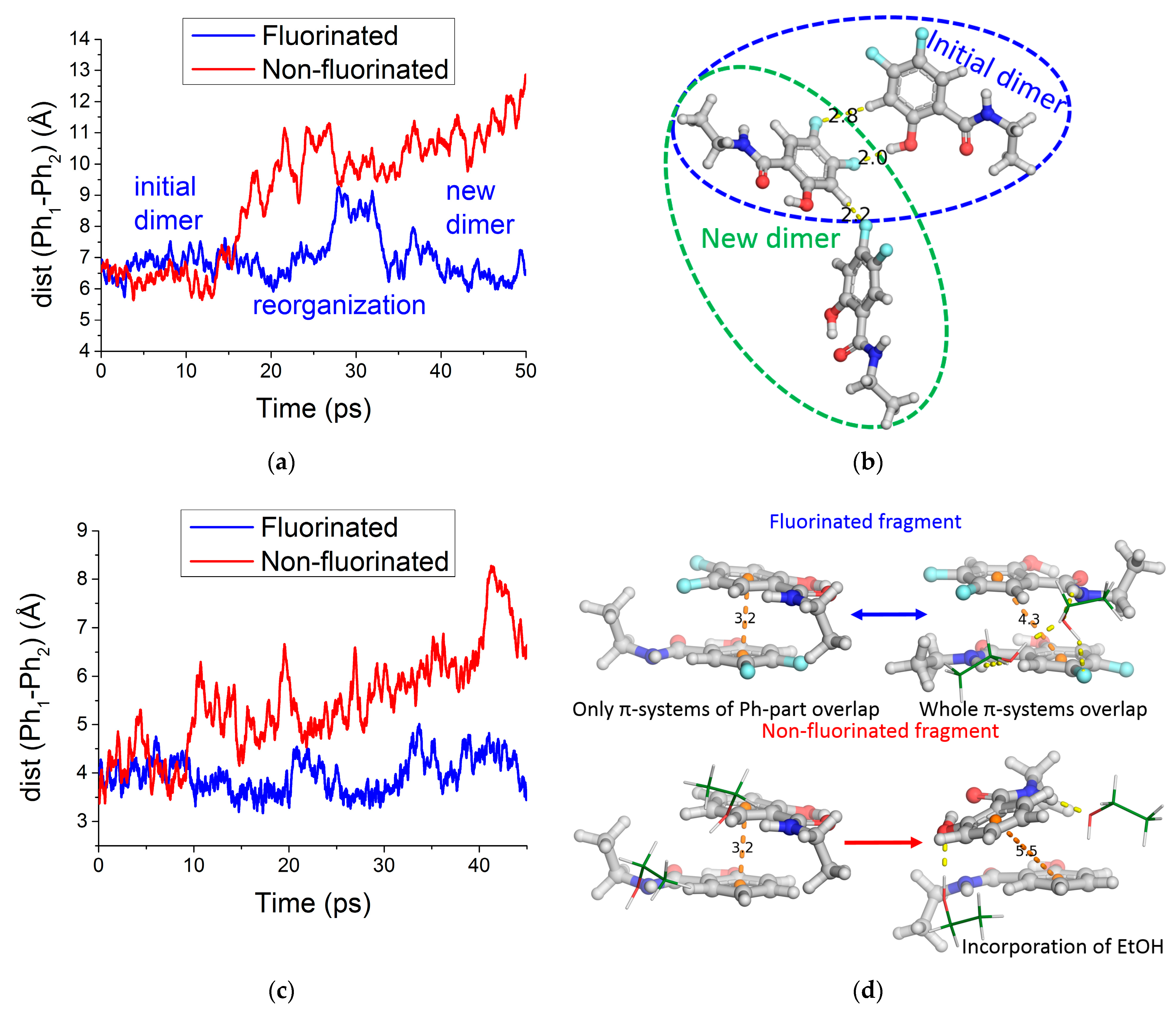
2.2.9. MD Simulations with QM/MM Potentials
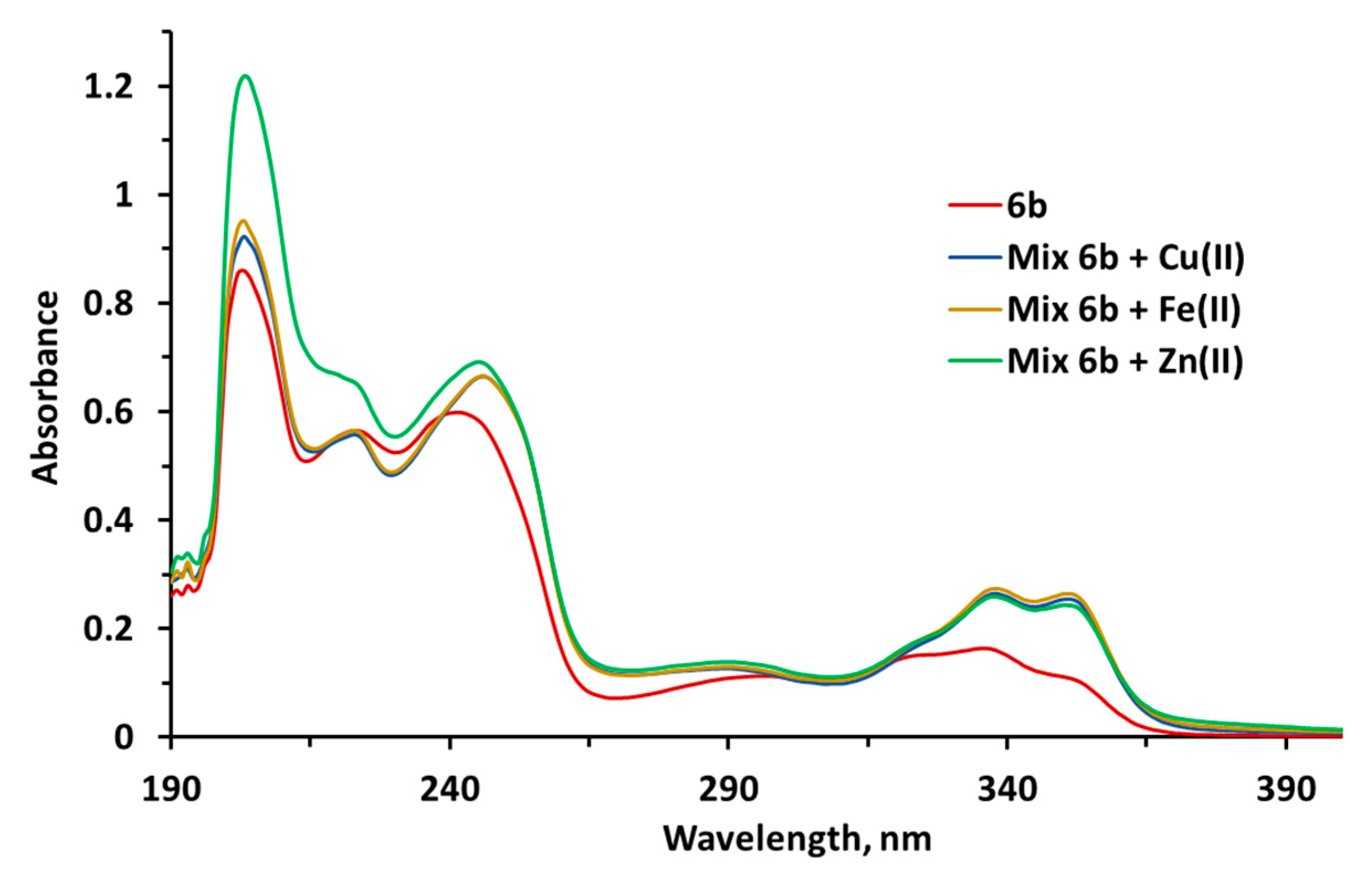
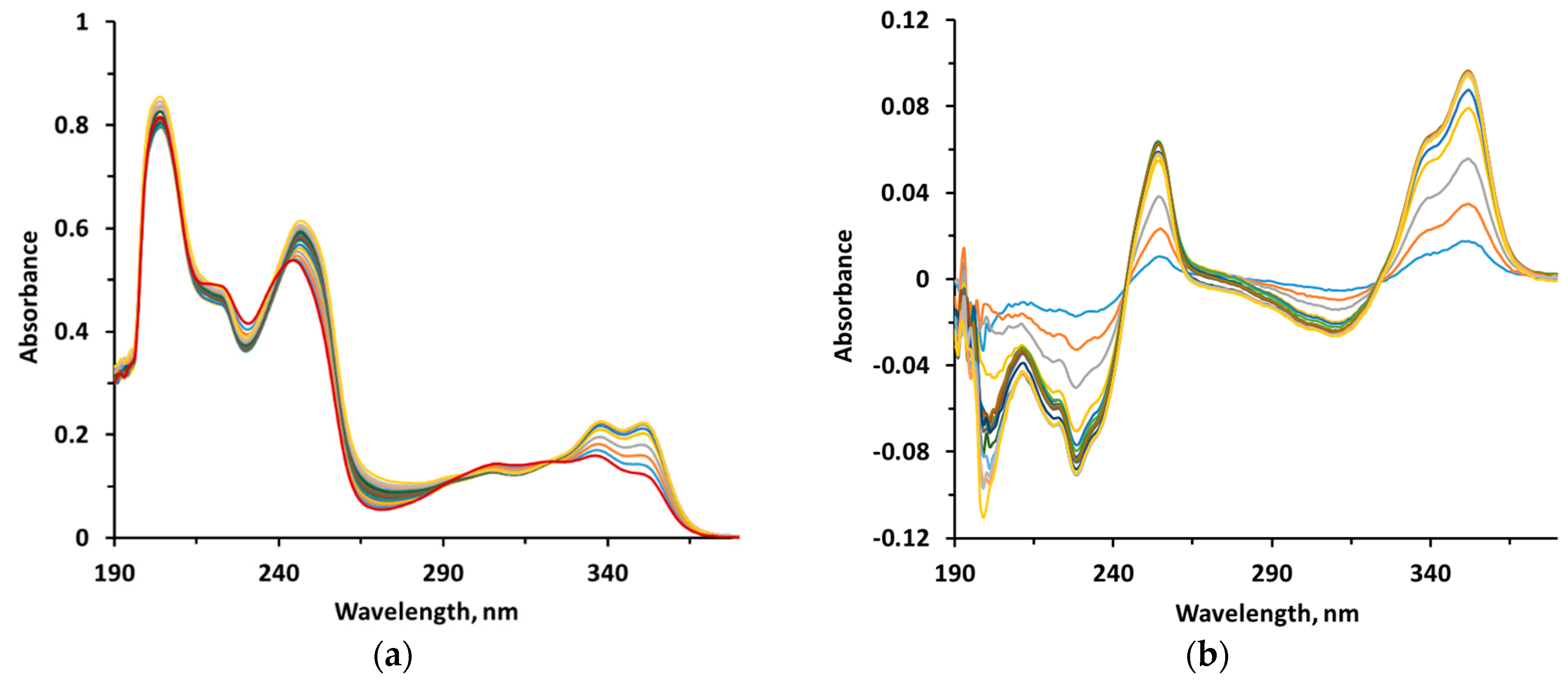
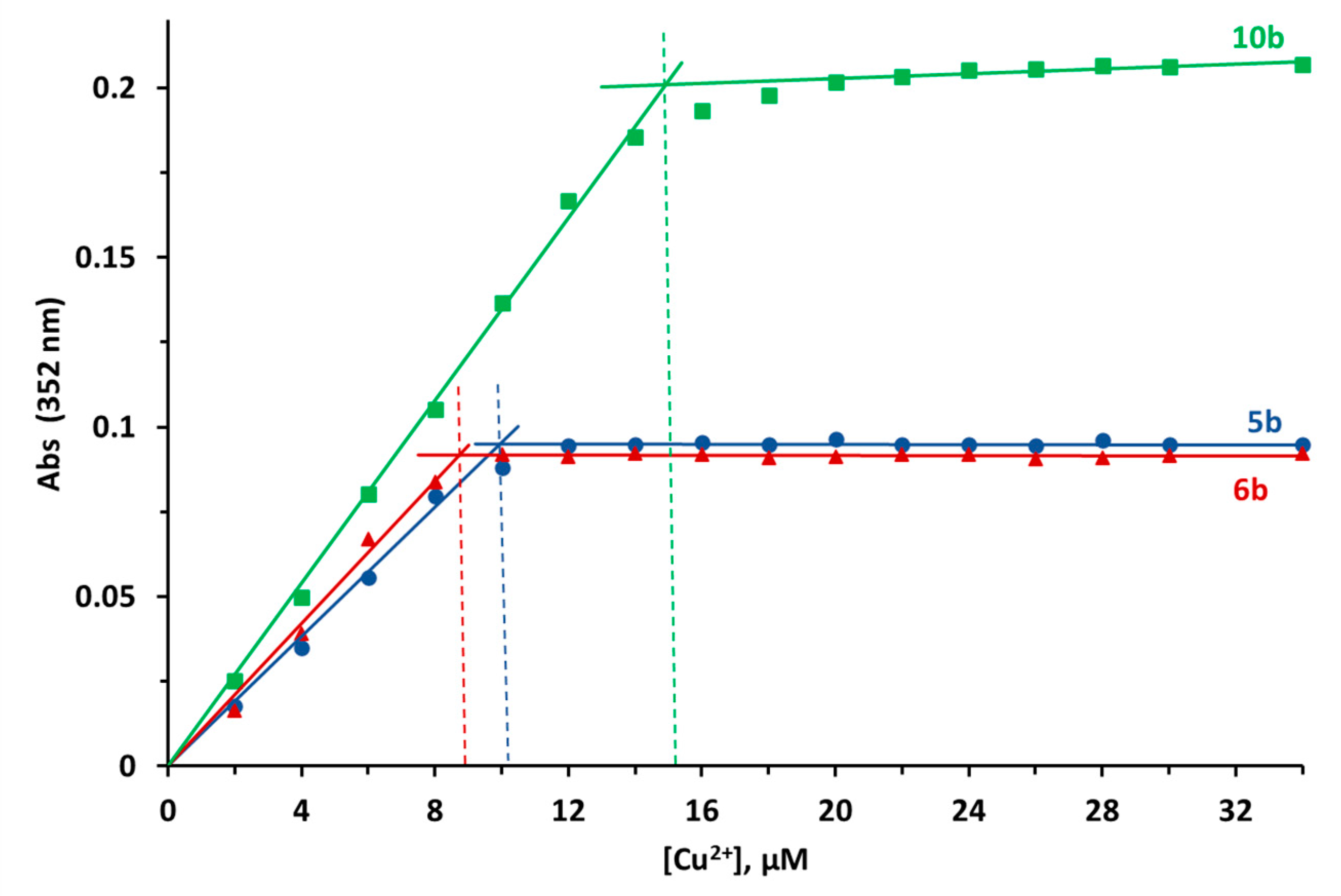
2.2.10. Metal-Chelating Properties
2.2.11. Cytotoxicity of Hybrids and Tacrine: MTT Assay in Primary Mouse Hepatocytes
2.2.12. Prediction of ADMET, Physicochemical, and PAINS Profiles
3. Experimental Section
3.1. Chemistry
3.1.1. Synthesis of Compounds 3a–c (General Procedure)
3.1.2. General Procedure for the Synthesis of Compounds 5–8
3.1.3. General Procedure of the Synthesis of Compounds 10a–c
3.2. Biological Investigations
3.2.1. Enzymatic Assays
In Vitro AChE, BChE, and CES Inhibition
Kinetic Study of AChE and BChE Inhibition: Determination of Steady-State Inhibition Constants
3.2.2. Propidium Iodide Displacement Studies
3.2.3. Inhibition of β-Amyloid (1–42) (Aβ42) Self-Aggregation
3.2.4. Antioxidant Activity
ABTS Radical Cation Scavenging Activity Assay
FRAP Assay
3.2.5. Metal Chelating Properties
3.3. Statistical Analyses
3.4. Molecular Modeling Studies
3.4.1. Preparation of the Molecules
3.4.2. Molecular Docking
3.4.3. Quantum Chemical Analysis of AO Activity
3.4.4. Prediction of ADMET, Physicochemical, and PAINS Profiles
3.5. Cytotoxicity Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer Disease. JAMA Neurol. 2016, 73, 867. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Liu, F.; Li, S.; Shi, D. Rational Multitargeted Drug Design Strategy from the Perspective of a Medicinal Chemist. J. Med. Chem. 2021, 64, 10581–10605. [Google Scholar] [CrossRef]
- Martinez, A.; Castro, A. Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2006, 15, 1–12. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Alkam, T.; Nitta, A.; Matsuyama, A.; Mizoguchi, H.; Suzuki, K.; Moussaoui, S.; Yu, Q.-S.; Greig, N.H.; Nagai, T.; et al. Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice. Behav. Brain Res. 2011, 225, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15, PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2005, 9, 101–124. [Google Scholar] [CrossRef]
- Hardy, J.; Bogdanovic, N.; Winblad, B.; Portelius, E.; Andreasen, N.; Cedazo-Minguez, A.; Zetterberg, H. Pathways to Alzheimer’s disease. J. Intern. Med. 2014, 275, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Selkoe, D.J. Soluble Oligomers of the Amyloid β-Protein: Impair Synaptic Plasticity and Behavior. In Synaptic Plasticity and the Mechanism of Alzheimer’s Disease; Selkoe, D.J., Triller, A., Christen, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 89–102. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, M.; Xu, S.; Wang, Y.; An, P.; Feng, C.; Chen, X.; Wei, X.; Han, Y.; Wang, Q. Bis(propyl)-cognitin Prevents beta-amyloid-induced Memory Deficits as Well as Synaptic Formation and Plasticity Impairments via the Activation of PI3-K Pathway. Mol. Neurobiol. 2016, 53, 3832–3841. [Google Scholar] [CrossRef]
- Hu, S.; Xian, Y.; Fan, Y.; Mak, S.; Wang, J.; Tang, J.; Pang, Y.; Pi, R.; Tsim, K.W.; Liu, F.; et al. Significant combination of Aβ aggregation inhibitory and neuroprotective properties in silico, in vitro and in vivo by bis(propyl)-cognitin, a multifunctional anti-Alzheimer’s agent. Eur. J. Pharmacol. 2020, 876, 173065. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- Moran, M.A.; Mufson, E.J.; Gomez-Ramos, P. Cholinesterases colocalize with sites of neurofibrillary degeneration in aged and Alzheimer’s brains. Acta Neuropathol. 1994, 87, 284–292. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Calderón, F. Acetylcholinesterase is a senile plaque component that promotes assembly of amyloid beta-peptide into Alzheimer’s filaments. Mol. Psychiatry 1996, 1, 359–361. [Google Scholar] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A Structural Motif of Acetylcholinesterase That Promotes Amyloid β-Peptide Fibril Formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Arce, M.P.; Rodríguez-Franco, M.I.; González-Muñoz, G.C.; Pérez, C.; López, B.; Villarroya, M.; López, M.G.; García, A.G.; Conde, S. Neuroprotective and Cholinergic Properties of Multifunctional Glutamic Acid Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2009, 52, 7249–7257. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Formosa, X.; Galdeano, C.; Gómez, T.; Muñoz-Torrero, D.; Ramírez, L.; Viayna, E.; Gómez, E.; Isambert, N.; Lavilla, R.; et al. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-Alzheimer drug candidates. Chem. Biol. Interact. 2010, 187, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.; Geula, C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 1994, 36, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Swaminathan, S.; Shen, L.; Foroud, T.M.; Hakonarson, H.; Huentelman, M.J.; Aisen, P.S.; et al. APOE and BCHE as modulators of cerebral amyloid deposition: A florbetapir PET genome-wide association study. Mol. Psychiatry 2014, 19, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Darvesh, S.; Reid, G.A. Reduced fibrillar β-amyloid in subcortical structures in a butyrylcholinesterase-knockout Alzheimer disease mouse model. Chem. Biol. Interact. 2016, 259, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimers Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Pohanka, M. Oxidative stress in Alzheimer disease as a target for therapy. Bratisl. Med. J. 2018, 119, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R.; Shenk, J.C.; Nunomura, A.; Smith, M.A.; Zhu, X.; Perry, G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol. Disord. Drug Targets 2008, 7, 3–10. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Sinha, M.; Thakurta, I.; Banerjee, P.; Chattopadhyay, M. Oxidative Stress and Amyloid Beta Toxicity in Alzheimer’s Disease: Intervention in a Complex Relationship by Antioxidants. Curr. Med. Chem. 2013, 20, 4648–4664. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s Diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Bush, A.I. Drug Development Based on the Metals Hypothesis of Alzheimer’s Disease. J. Alzheimers Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Strodel, B.; Coskuner-Weber, O. Transition Metal Ion Interactions with Disordered Amyloid-beta Peptides in the Pathogenesis of Alzheimer’s Disease: Insights from Computational Chemistry Studies. J. Chem. Inf. Model. 2019, 59, 1782–1805. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.-S.; Wang, X.-B.; Li, J.-Y.; Yang, L.; Kong, L.-Y. Design, synthesis and evaluation of novel tacrine–coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur. J. Med. Chem. 2013, 64, 540–553. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in multifunctional metal chelators as potential drugs for Alzheimer’s disease. Coord. Chem. Rev. 2016, 327–328, 287–303. [Google Scholar] [CrossRef]
- Mishra, P.; Kumar, A.; Panda, G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Milelli, A.; De Simone, A.; Ticchi, N.; Chen, H.H.; Betari, N.; Andrisano, V.; Tumiatti, V. Tacrine-based Multifunctional Agents in Alzheimer’s Disease: An Old Story in Continuous Development§. Curr. Med. Chem. 2017, 24, 3522–3546. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer’s disease. Curr. Top. Med. Chem. 2017, 17, 1006–1026. [Google Scholar] [CrossRef]
- Przybyłowska, M.; Kowalski, S.; Dzierzbicka, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Tacrine Analogues. Curr. Neuropharmacol. 2019, 17, 472–490. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer’s disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef] [PubMed]
- Minarini, A.; Milelli, A.; Simoni, E.; Rosini, M.; Bolognesi, M.; Marchetti, C.; Tumiatti, V. Multifunctional Tacrine Derivatives in Alzheimer’s Disease. Curr. Top. Med. Chem. 2013, 13, 1771–1786. [Google Scholar] [CrossRef] [PubMed]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 128, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent Development of Multifunctional Agents as Potential Drug Candidates for the Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2014, 22, 373–404. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Chadha, N.; Silakari, O. Hybrids: A new paradigm to treat Alzheimer’s disease. Mol. Divers. 2016, 20, 271–297. [Google Scholar] [CrossRef]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational Approach To Discover Multipotent Anti-Alzheimer Drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef]
- Xie, S.-S.; Lan, J.-S.; Wang, X.-B.; Jiang, N.; Dong, G.; Li, Z.-R.; Wang, K.D.G.; Guo, P.-P.; Kong, L.-Y. Multifunctional tacrine–trolox hybrids for the treatment of Alzheimer’s disease with cholinergic, antioxidant, neuroprotective and hepatoprotective properties. Eur. J. Med. Chem. 2015, 93, 42–50. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine−8-Hydroxyquinoline Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Neuroprotective, Cholinergic, Antioxidant, and Copper-Complexing Properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine–Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Faingold, I.I.; Poletaeva, D.A.; Soldatova, Y.V.; Kotelnikova, R.A.; Serkov, I.V.; et al. New Multifunctional Agents Based on Conjugates of 4-Amino-2,3-polymethylenequinoline and Butylated Hydroxytoluene for Alzheimer’s Disease Treatment. Molecules 2020, 25, 5891. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Serkov, I.V.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Kochetkova, E.A.; Proshin, A.N.; Bachurin, S.O. Novel conjugates of 4-amino-2,3-polymethylenequinolines and vanillin as potential multitarget agents for AD treatment. Mendeleev Commun. 2021, 31, 606–608. [Google Scholar] [CrossRef]
- Mao, F.; Huang, L.; Luo, Z.; Liu, A.; Lu, C.; Xie, Z.; Li, X. O-Hydroxyl- or o-amino benzylamine-tacrine hybrids: Multifunctional biometals chelators, antioxidants, and inhibitors of cholinesterase activity and amyloid-β aggregation. Bioorg. Med. Chem. 2012, 20, 5884–5892. [Google Scholar] [CrossRef]
- Borges, R.S.; Pereira, G.A.N.; Vale, J.K.L.; França, L.C.S.; Monteiro, M.C.; Alves, C.N.; da Silva, A.B.F. Design and Evaluation of 4-Aminophenol and Salicylate Derivatives as Free-Radical Scavenger. Chem. Biol. Drug Des. 2013, 81, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S.; Castle, S.L. The antioxidant properties of salicylate derivatives: A possible new mechanism of anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2015, 25, 4808–4811. [Google Scholar] [CrossRef]
- Song, Q.; Li, Y.; Cao, Z.; Qiang, X.; Tan, Z.; Deng, Y. Novel salicylamide derivatives as potent multifunctional agents for the treatment of Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2019, 84, 137–149. [Google Scholar] [CrossRef]
- Grishchenko, M.V.; Makhaeva, G.F.; Burgart, Y.V.; Rudakova, E.V.; Boltneva, N.P.; Kovaleva, N.V.; Serebryakova, O.G.; Lushchekina, S.V.; Astakhova, T.Y.; Zhilina, E.F.; et al. Conjugates of Tacrine with Salicylamide as Promising Multitarget Agents for Alzheimer’s Disease. ChemMedChem 2022, 17, e202200080. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, 121, 4678–4742. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Dolbier, W.R. Fluorine chemistry at the millennium. J. Fluor. Chem. 2005, 126, 157–163. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluor. Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Luo, W.; Li, Y.P.; He, Y.; Huang, S.L.; Li, D.; Gu, L.Q.; Huang, Z.S. Synthesis and evaluation of heterobivalent tacrine derivatives as potential multi-functional anti-Alzheimer agents. Eur. J. Med. Chem. 2011, 46, 2609–2616. [Google Scholar] [CrossRef]
- Carlier, P.R.; Chow, E.S.H.; Han, Y.; Liu, J.; Yazal, J.E.; Pang, Y.-P. Heterodimeric Tacrine-Based Acetylcholinesterase Inhibitors: Investigating Ligand−Peripheral Site Interactions. J. Med. Chem. 1999, 42, 4225–4231. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Rudakova, E.V.; Kovaleva, N.V.; Lushchekina, S.V.; Boltneva, N.P.; Proshin, A.N.; Shchegolkov, E.V.; Burgart, Y.V.; Saloutin, V.I. Cholinesterase and carboxylesterase inhibitors as pharmacological agents. Russ. Chem. Bull. 2019, 68, 967–984. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Rudakova, E.V.; Serebryakova, O.G.; Aksinenko, A.Y.; Lushchekina, S.V.; Bachurin, S.O.; Richardson, R.J. Esterase profiles of organophosphorus compounds in vitro predict their behavior in vivo. Chem. Biol. Interact. 2016, 259, 332–342. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Boltneva, N.P.; Lushchekina, S.V.; Serebryakova, O.G.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Bachurin, S.O.; Richardson, R.J. Synthesis, molecular docking and biological evaluation of N,N-disubstituted 2-aminothiazolines as a new class of butyrylcholinesterase and carboxylesterase inhibitors. Bioorg. Med. Chem. 2016, 24, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Sokolov, V.B.; Grigoriev, V.V.; Serebryakova, O.G.; Vikhareva, E.A.; Aksinenko, A.Y.; Barreto, G.E.; Aliev, G.; et al. Conjugates of γ-carbolines and phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer disease. Sci. Rep. 2015, 5, 13164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhaeva, G.F.; Radchenko, E.V.; Palyulin, V.A.; Rudakova, E.V.; Aksinenko, A.Y.; Sokolov, V.B.; Zefirov, N.S.; Richardson, R.J. Organophosphorus compound esterase profiles as predictors of therapeutic and toxic effects. Chem. Biol. Interact. 2013, 203, 231–237. [Google Scholar] [CrossRef]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Tsurkan, L.G.; Hatfield, M.J.; Edwards, C.C.; Hyatt, J.L.; Potter, P.M. Inhibition of human carboxylesterases hCE1 and hiCE by cholinesterase inhibitors. Chem. Biol. Interact. 2013, 203, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Lushchekina, S.V.; Kots, E.D.; Novichkova, D.A.; Petrov, K.A.; Masson, P. Role of acetylcholinesterase in β-amyloid aggregation studied by accelerated molecular dynamics. BioNanoScience 2016, 7, 396–402. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Makhaeva, G.F.; Shevtsova, E.F.; Aksinenko, A.Y.; Grigoriev, V.V.; Shevtsov, P.N.; Goreva, T.V.; Epishina, T.A.; Kovaleva, N.V.; Pushkareva, E.A.; et al. Conjugation of Aminoadamantane and γ-Carboline Pharmacophores Gives Rise to Unexpected Properties of Multifunctional Ligands. Molecules 2021, 26, 5527. [Google Scholar] [CrossRef]
- Ghotbi, G.; Mahdavi, M.; Najafi, Z.; Moghadam, F.H.; Hamzeh-Mivehroud, M.; Davaran, S.; Dastmalchi, S. Design, synthesis, biological evaluation, and docking study of novel dual-acting thiazole-pyridiniums inhibiting acetylcholinesterase and beta-amyloid aggregation for Alzheimer’s disease. Bioorg. Chem. 2020, 103, 104186. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Montero, J.M.; Agis-Torres, A.; Solano, D.; Sollhuber, M.; Fernandez, M.; Villaro, W.; Gomez-Canas, M.; Garcia-Arencibia, M.; Fernandez-Ruiz, J.; Egea, J.; et al. Analogues of cannabinoids as multitarget drugs in the treatment of Alzheimer’s disease. Eur. J. Pharmacol. 2021, 895, 173875. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Ruiz, P.; Rubio, L.; Garcia-Palomero, E.; Dorronsoro, I.; del Monte-Millan, M.; Valenzuela, R.; Usan, P.; de Austria, C.; Bartolini, M.; Andrisano, V.; et al. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: New disease-modifying agents for Alzheimer’s disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar] [CrossRef]
- Bartolini, M.; Naldi, M.; Fiori, J.; Valle, F.; Biscarini, F.; Nicolau, D.V.; Andrisano, V. Kinetic characterization of amyloid-beta 1–42 aggregation with a multimethodological approach. Anal. Biochem. 2011, 414, 215–225. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef] [PubMed]
- Safarizadeh, H.; Garkani-Nejad, Z. Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer’s amyloid-beta aggregation: Insight into mechanism of interactions and parameters for design of new inhibitors. J. Mol. Graph. Model. 2019, 87, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Jokar, S.; Erfani, M.; Bavi, O.; Khazaei, S.; Sharifzadeh, M.; Hajiramezanali, M.; Beiki, D.; Shamloo, A. Design of peptide-based inhibitor agent against amyloid-beta aggregation: Molecular docking, synthesis and in vitro evaluation. Bioorg. Chem. 2020, 102, 104050. [Google Scholar] [CrossRef]
- Giulian, D.; Haverkamp, L.J.; Yu, J.; Karshin, W.; Tom, D.; Li, J.; Kazanskaia, A.; Kirkpatrick, J.; Roher, A.E. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J. Biol. Chem. 1998, 273, 29719–29726. [Google Scholar] [CrossRef] [Green Version]
- Ariga, T.; Miyatake, T.; Yu, R.K. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: Amyloidogenesis and therapeutic strategies—A review. J. Neurosci. Res. 2010, 88, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Elkina, N.A.; Shchegolkov, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Serebryakova, O.G.; Rudakova, E.V.; Kovaleva, N.V.; Radchenko, E.V.; Palyulin, V.A.; et al. Synthesis, molecular docking, and biological evaluation of 3-oxo-2-tolylhydrazinylidene-4,4,4-trifluorobutanoates bearing higher and natural alcohol moieties as new selective carboxylesterase inhibitors. Bioorg. Chem. 2019, 91, 103097. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.H.; Ji, C. MolGpka: A Web Server for Small Molecule pKa Prediction Using a Graph-Convolutional Neural Network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef] [Green Version]
- Amic, D.; Stepanic, V.; Lucic, B.; Markovic, Z.; Dimitric Markovic, J.M. PM6 study of free radical scavenging mechanisms of flavonoids: Why does O-H bond dissociation enthalpy effectively represent free radical scavenging activity? J. Mol. Model. 2013, 19, 2593–2603. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Yadav, H.R.; Choudhury, A.R. Can C H⋯F C hydrogen bonds alter crystal packing features in the presence of N H⋯O C hydrogen bond? J. Biomol. Struct. 2017, 1150, 469–480. [Google Scholar] [CrossRef]
- Cole, J.C.; Taylor, R. Intermolecular Interactions of Organic Fluorine Seen in Perspective. Cryst. Growth Des. 2022, 22, 1352–1364. [Google Scholar] [CrossRef]
- Perez, C.A.; Wei, Y.; Guo, M. Iron-binding and anti-Fenton properties of baicalein and baicalin. J. Inorg. Biochem. 2009, 103, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-L.; Cai, P.; Liu, Q.-H.; Yang, X.-L.; Fang, S.-Q.; Tang, Y.-W.; Wang, C.; Wang, X.-B.; Kong, L.-Y. Design, synthesis, and evaluation of salicyladimine derivatives as multitarget-directed ligands against Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 5917–5928. [Google Scholar] [CrossRef]
- Lou, Y.-H.; Wang, J.-S.; Dong, G.; Guo, P.-P.; Wei, D.-D.; Xie, S.-S.; Yang, M.-H.; Kong, L.-Y. The acute hepatotoxicity of tacrine explained by 1H NMR based metabolomic profiling. Toxicol. Res. 2015, 4, 1465–1478. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Deng, X.; Wang, Z.; Liu, J.; Xiong, S.; Xiong, R.; Cao, X.; Chen, Y.; Zheng, X.; Tang, G. Design, synthesis and biological evaluation of flavonoid salicylate derivatives as potential anti-tumor agents. RSC Adv. 2017, 7, 38171–38178. [Google Scholar] [CrossRef] [Green Version]
- Shchegol’kov, E.V.; Shchur, I.V.; Burgart, Y.V.; Saloutin, V.I.; Solodnikov, S.Y.; Krasnykh, O.P.; Kravchenko, M.A. A convenient and efficient approach to polyfluorosalicylic acids and their tuberculostatic activity. Bioorg. Med. Chem. Lett. 2016, 26, 2455–2458. [Google Scholar] [CrossRef]
- Zhang, H.; Peng, Y.; Zhuo, L.; Wang, Y.; Zeng, G.; Wang, S.; Long, L.; Li, X.; Wang, Z. Recent advance on pleiotropic cholinesterase inhibitors bearing amyloid modulation efficacy. Eur. J. Med. Chem. 2022, 242, 114695. [Google Scholar] [CrossRef]
- Gniazdowska, E.; Koźmiński, P.; Wasek, M.; Bajda, M.; Sikora, J.; Mikiciuk-Olasik, E.; Szymański, P. Synthesis, physicochemical and biological studies of technetium-99m labeled tacrine derivative as a diagnostic tool for evaluation of cholinesterase level. Bioorg. Med. Chem. 2017, 25, 912–920. [Google Scholar] [CrossRef]
- Cheng, Z.-Q.; Zhu, K.-K.; Zhang, J.; Song, J.-L.; Muehlmann, L.A.; Jiang, C.-S.; Liu, C.-L.; Zhang, H. Molecular-docking-guided design and synthesis of new IAA-tacrine hybrids as multifunctional AChE/BChE inhibitors. Bioorg. Chem. 2019, 83, 277–288. [Google Scholar] [CrossRef]
- Bornstein, J.J.; Eckroat, T.J.; Houghton, J.L.; Jones, C.K.; Green, K.D.; Garneau-Tsodikova, S. Tacrine-mefenamic acid hybrids for inhibition of acetylcholinesterase. MedChemComm 2011, 2, 406–412. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Rudakova, E.V.; Proshin, A.N.; Serkov, I.V.; Radchenko, E.V.; Palyulin, V.A.; et al. New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer’s Disease Treatment. Molecules 2020, 25, 3915. [Google Scholar] [CrossRef]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. Site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Makhaeva, G.F.; Shevtsova, E.F.; Boltneva, N.P.; Kovaleva, N.V.; Lushchekina, S.V.; Rudakova, E.V.; Dubova, L.G.; Vinogradova, D.V.; Sokolov, V.B.; et al. Conjugates of methylene blue with gamma-carboline derivatives as new multifunctional agents for the treatment of neurodegenerative diseases. Sci. Rep. 2019, 9, 4873. [Google Scholar] [CrossRef] [Green Version]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Löwdin, P.-O. On the nonorthogonality problem. In Advances in Quantum Chemistry; Per-Olov, L., Ed.; Academic Press: New York, NY, USA; London, UK, 1970; Volume 5, pp. 185–199. [Google Scholar]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDock Tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16 Revision C. 01. 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila-Schumacher, S.I.L.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. TeraChem: A graphical processing unit—Accelerated electronic structure package for large-scale ab initio molecular dynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 11, e1494. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Vermaas, J.V.; Crowley, M.F.; Beckham, G.T. Molecular Lignin Solubility and Structure in Organic Solvents. ACS Sustain. Chem. Eng. 2020, 8, 17839–17850. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided. Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Dyabina, A.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Prediction of blood-brain barrier permeability of organic compounds. Dokl. Biochem. Biophys. 2016, 470, 371–374. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A. Towards Deep Neural Network Models for the Prediction of the Blood-Brain Barrier Permeability for Diverse Organic Compounds. Molecules 2020, 25, 5901. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.A.; Zefirov, N.S. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef] [PubMed]
- ADMET Prediction Service. Available online: http://qsar.chem.msu.ru/admet/ (accessed on 15 June 2022).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RDKit: Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 15 June 2022).
- Charni-Natan, M.; Goldstein, I. Protocol for Primary Mouse Hepatocyte Isolation. STAR Protoc. 2020, 1, 100086. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibitory Activity against AChE, BChE, and CES IC50 (µM) or Inhibition at 20 µM (%) 1 | Propidium Displacement from EeAChE PAS, (%) 1 | Inhibition of Aβ42 Self- Aggregation, (%) 2 | ||||
|---|---|---|---|---|---|---|---|
| No |  Where R | n | Human Erythrocyte AChE | Equine Serum BChE | Porcine Liver CES 1 | ||
| Salicylamides | |||||||
| 5a |  | 4 | 1.17 ± 0.05 | 0.0107 ± 0.0005 | 1.6 ± 1.1% | 11.8 ± 0.9 | 20.7 ± 1.6 |
| 5b | 6 | 0.294 ± 0.008 | 0.0119 ± 0.0001 | 19.5 ± 0.9% | 12.6 ± 1.0 | 41.5 ± 2.9 | |
| 5c | 8 | 0.224 ± 0.017 | 0.0104 ± 0.0013 | 29.5 ± 2.5% | 14.2 ± 1.1 | 71.9 ± 5.7 | |
| Methoxybenzoylamides | |||||||
| 6a |  | 4 | 1.92 ± 0.13 | 0.0105 ± 0.0007 | 3.4 ± 0.7% | 10.2 ± 0.9 | 12.3 ± 0.9 |
| 6b | 6 | 0.646 ± 0.041 | 0.0377 ± 0.0031 | 8.0 ± 1.3% | 12.5 ± 0.8 | 30.8 ± 2.1 | |
| 6c | 8 | 0.150 ± 0.011 | 0.0475 ± 0.0032 | 10.6 ± 1.2% | 14.1 ± 0.9 | 55.6 ± 4.4 | |
| Difluorosalicylamides | |||||||
| 7a |  | 4 | 0.500 ± 0.030 | 0.0291 ± 0.0022 | 4.3 ± 0.9% | 10.4 ± 0.9 | 17.4 ± 1.2 |
| 7b | 6 | 0.249 ± 0.017 | 0.0317 ± 0.0030 | 7.1 ± 1.0% | 12.2 ± 0.9 | 24.1 ± 2.1 | |
| 7c | 8 | 0.188 ± 0.015 | 0.0226 ± 0.0018 | 8.6 ± 0.7% | 13.6 ± 1.0 | 37.2 ± 2.9 | |
| Difluoromethoxybenzoylamides | |||||||
| 8a |  | 4 | 1.22 ± 0.07 | 0.0547 ± 0.0034 | 6.5 ± 1.1% | 13.2 ± 1.0 | 10.4 ± 0.9 |
| 8b | 6 | 0.712 ± 0.067 | 0.0702 ± 0.0042 | 6.3 ± 0.9% | 13.8 ± 0.9 | 29.7 ± 2.3 | |
| 8c | 8 | 0.142 ± 0.011 | 0.0317 ± 0.0024 | 19.8 ± 3.1% | 15.1 ± 1.2 | 54.1 ± 4.3 | |
| Salicylimines | |||||||
| 10a |  | 4 | 0.743 ± 0.044 | 0.127 ± 0.006 | 20.8 ± 0.4% | 18.6 ± 1.6 | 57.9 ± 4.6 |
| 10b | 6 | 0.103 ± 0.006 | 0.0354 ± 0.0017 | 17.9 ± 0.8% | 18.2 ± 1.4 | 67.9 ± 5.4 | |
| 10c | 8 | 0.0826 ± 0.0046 | 0.0156 ± 0.0012 | 23.0 ± 0.3% | 19.8 ± 1.7 | 80.1 ± 5.6 | |
N-hexylsalicylamide [59] | n.a. | 10.6 ± 0.8% | 4.9 ± 1.0% | n.d. | n.a | ||
| Salicylic acid | n.a. | 47.6 ± 3.2 | 8.9 ± 0.6% | n.d. | 6.5 ± 1.2 | ||
| Tacrine | 0.601 ± 0.047 | 0.0295 ± 0.0020 | n.a. | 3.1 ± 0.2 | 5.9 ± 0.5 | ||
| BNPP | n.a. | n.a. | 1.80 ± 0.11 | n.d. | n.d. | ||
| Donepezil | 0.040 ± 0.004 | 19.2 ± 3.0 | n.a. | 11.9 ± 0.9 | n.d. | ||
| Myricetin | n.d. | n.d. | n.d. | n.d. | 74.7 ± 5.2 | ||
| Propidium iodide | n.d. | n.d. | n.d. | n.d. | 90.7 ± 7.1 | ||
| Compound | AChE | eqBChE | ||
|---|---|---|---|---|
| Ki (µM) | αKi (µM) | Ki (µM) | αKi (µM) | |
| 5c | 0.152 ± 0.012 | 0.263 ± 0.018 | 0.0106 ± 0.0007 | 0.0171 ± 0.0013 |
| 6c | 0.104 ± 0.004 | 0.213 ± 0.012 | 0.0188 ± 0.0011 | 0.0314 ± 0.0025 |
| 7c | 0.125 ± 0.003 | 0.134 ± 0.009 | 0.0173 ± 0.0006 | 0.0243 ± 0.0001 |
| 8c | 0.0536 ± 0.0025 | 0.0998 ± 0.0058 | 0.0177 ± 0.0007 | 0.0187 ± 0.0013 |
| 10c | 0.0299 ± 0.0021 | 0.0644 ± 0.0040 | 0.0084 ± 0.0006 | 0.0152 ± 0.0009 |
| Compound | ABTS•+ Scavenging Activity | ||||
|---|---|---|---|---|---|
| No |  Where R | n | TEAC 1 | IC50, μM 2 | FRAP, TE 3 |
| Salicylamides | |||||
| 5a |  | 4 | 0.73 ± 0.04 | 25.3 ± 1.1 | 0.02 |
| 5b | 6 | 0.90 ± 0.05 | 21.3 ± 1.5 | n.a. | |
| 5c | 8 | 0.75 ± 0.04 | 25.5 ± 1.1 | n.a. | |
| Methoxybenzoylamides | |||||
| 6a |  | 4 | 0.02 (n.a.) | n.d. | 0.03 |
| 6b | 6 | n.a. | n.d. | 0.02 | |
| 6c | 8 | n.a. | n.d. | n.a. | |
| Difluorosalicylamides | |||||
| 7a |  | 4 | 0.060 ± 0.002 | >200 | n.a. |
| 7b | 6 | 0.030 ± 0.001 | >200 | 0.03 | |
| 7c | 8 | 0.040 ± 0.002 | >200 | 0.06 | |
| Difluoromethoxybenzoylamides | |||||
| 8a |  | 4 | n.a. | n.d. | 0.03 |
| 8b | 6 | n.a. | n.d. | 0.02 | |
| 8c | 8 | n.a. | n.d. | 0.02 | |
| Salicylimines | |||||
| 10a |  | 4 | 0.42 ± 0.03 | 35.8 ± 1.9 | 0.02 |
| 10b | 6 | 0.47 ± 0.04 | 31.8 ± 1.6 | 0.03 | |
| 10c | 8 | 0.42 ± 0.04 | 34.2 ± 1.8 | n.a. | |
N-hexylsalicylamide [59] | 0.39 ± 0.04 | 46.5 ± 2.3 | n.d. | ||
| Salicylic acid | n.a. | n.a. | 0.02 | ||
| Tacrine | n.a. | n.a. | n.a. | ||
| Trolox | 1.00 | 19.8 ± 0.8 | 1.00 | ||
| Ascorbic acid | 0.96 ± 0.03 | 22.6 ± 1.2 | 1.28 ± 0.11 | ||
| Compound | Ethanol BDE, kcal/mol | Ethanol IP, kcal/mol | TEAC | IC50, μM |
|---|---|---|---|---|
| 5a | 81.7 | 124.7 | 0.72 ± 0.04 | 25.3 ± 1.1 |
| 10a | 84.7 | 126.1 | 0.42 ± 0.03 | 35.8 ± 1.9 |
| 6a | 88.9 | 124.3 | 0.02 (n.a.) | n.d. |
| 7a | 82.0 | 121.9 | 0.060 ± 0.002 | >200 |
| Sample | λmax, nm | Shift of Absorbance 1, nm | ||
|---|---|---|---|---|
| Cu(II) | Fe(II) | Zn(II) | ||
| 5b [59] | 337, 323, 308 | 352 | 351 | 351 |
| 237 | 254 | 254 | 254 | |
| 6b | 336 | 352 | 352 | 352 |
| 241 | 254 | 254 | 253 | |
| 7b | 337 | 352 | 352 | 352 |
| 246 | 253 | 253 | - | |
| 8b | 336 | 351 | 351 | 351 |
| 245 | 254 | 255 | 251 | |
| 10b | 323 | 352 | 352 | 352 |
| 246 | 255 | 254 | 254 | |
| Compound | IC50, μM (Mean ± SEM, n = 3) |
|---|---|
| 5b | 4.89 ± 0.08 |
| 6c | 3.81 ± 0.07 |
| 7b | 15.16 ± 0.09 |
| 8b | 3.95 ± 0.22 |
| 10c | 5.87 ± 0.14 |
| Tacrine | 3.78 ± 0.15 |
| Compound | MW | LogPow | pSaq | LogBB | HIA, % | hERG pKi | hERG pIC50 | QED |
|---|---|---|---|---|---|---|---|---|
| 5a | 389.50 | 4.38 | 5.51 | –0.12 | 95 | 4.86 | 6.03 | 0.52 |
| 5b | 417.55 | 5.14 | 6.12 | –0.13 | 95 | 4.79 | 6.15 | 0.42 |
| 5c | 445.61 | 5.80 | 6.80 | 0.25 | 95 | 5.18 | 6.60 | 0.32 |
| 6a | 403.53 | 4.62 | 5.82 | 0.23 | 100 | 4.86 | 6.43 | 0.53 |
| 6b | 431.58 | 5.36 | 6.46 | 0.23 | 100 | 4.79 | 6.56 | 0.41 |
| 6c | 459.63 | 6.02 | 7.11 | 0.59 | 100 | 5.18 | 7.03 | 0.31 |
| 7a | 425.48 | 4.51 | 5.90 | –0.03 | 100 | 4.93 | 5.63 | 0.48 |
| 7c | 481.59 | 5.97 | 7.50 | 0.34 | 100 | 5.25 | 6.11 | 0.28 |
| 7b | 453.53 | 5.26 | 6.77 | –0.04 | 100 | 4.87 | 5.73 | 0.37 |
| 8a | 439.51 | 4.75 | 6.18 | 0.06 | 100 | 4.93 | 5.74 | 0.48 |
| 8b | 467.56 | 5.52 | 7.03 | 0.05 | 100 | 4.87 | 5.84 | 0.37 |
| 8c | 495.62 | 6.19 | 7.77 | 0.43 | 100 | 5.25 | 6.22 | 0.28 |
| 10a | 373.50 | 4.97 | 5.83 | 0.02 | 89 | 5.16 | 5.76 | 0.45 |
| 10b | 401.55 | 5.61 | 6.63 | –0.07 | 89 | 5.08 | 6.16 | 0.35 |
| 10c | 429.61 | 6.19 | 7.31 | 0.31 | 89 | 5.44 | 6.59 | 0.26 |
| Tacrine | 198.27 | 2.95 | 1.52 | 0.00 | 93 | 4.98 | 4.98 | 0.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Grishchenko, M.V.; Lushchekina, S.V.; Astakhova, T.Y.; Serebryakova, O.G.; Timokhina, E.N.; Zhilina, E.F.; et al. Conjugates of Tacrine and Salicylic Acid Derivatives as New Promising Multitarget Agents for Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 2285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032285
Makhaeva GF, Kovaleva NV, Rudakova EV, Boltneva NP, Grishchenko MV, Lushchekina SV, Astakhova TY, Serebryakova OG, Timokhina EN, Zhilina EF, et al. Conjugates of Tacrine and Salicylic Acid Derivatives as New Promising Multitarget Agents for Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(3):2285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032285
Chicago/Turabian StyleMakhaeva, Galina F., Nadezhda V. Kovaleva, Elena V. Rudakova, Natalia P. Boltneva, Maria V. Grishchenko, Sofya V. Lushchekina, Tatiana Y. Astakhova, Olga G. Serebryakova, Elena N. Timokhina, Ekaterina F. Zhilina, and et al. 2023. "Conjugates of Tacrine and Salicylic Acid Derivatives as New Promising Multitarget Agents for Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 3: 2285. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032285