

The Role of Alpha-Fetoprotein (AFP) in Contemporary Oncology: The Path from a Diagnostic Biomarker to an Anticancer Drug
 , , ,
, , , 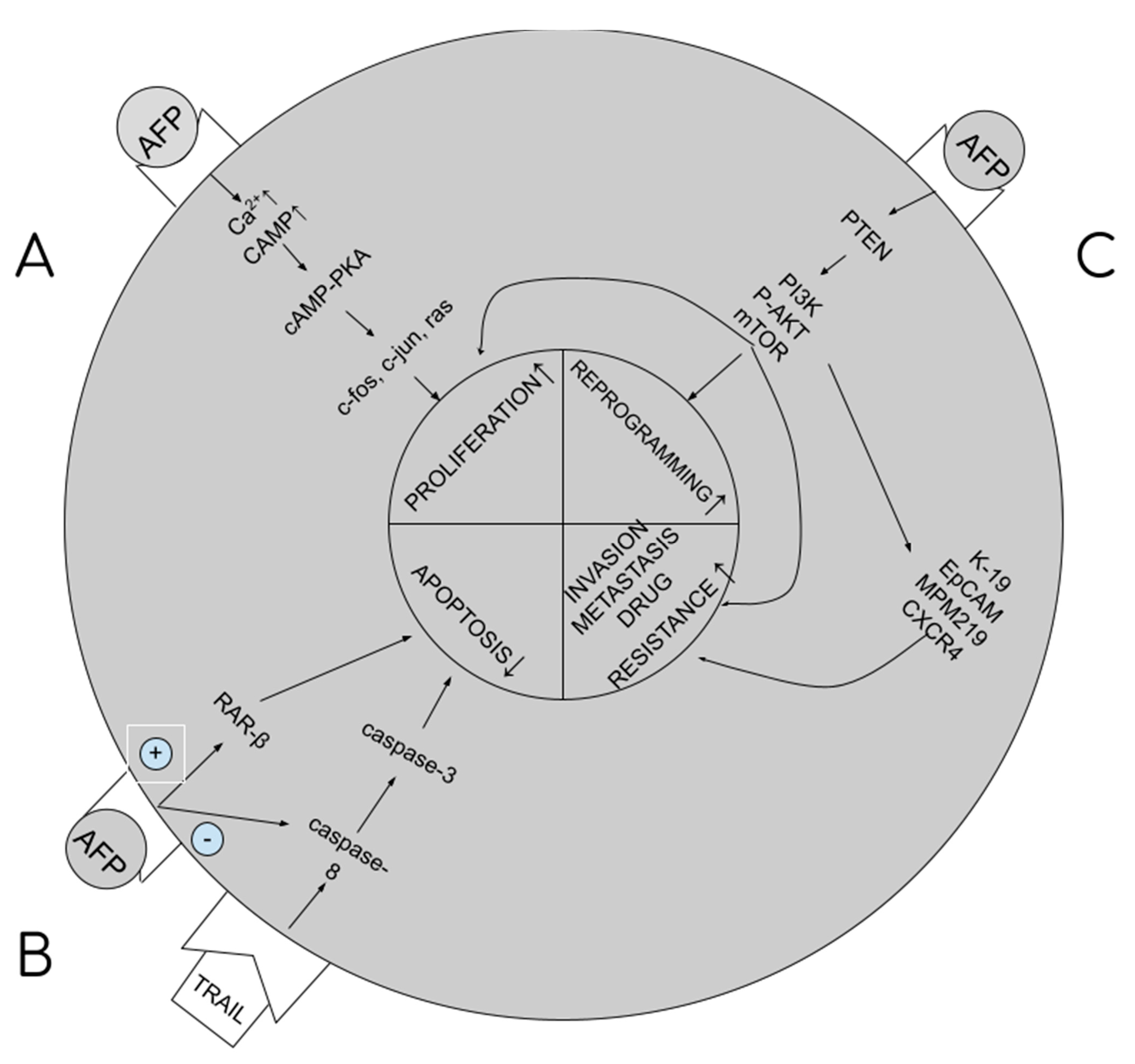{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of AFP in Carcinogenesis Process
3. AFP as an Oncological Carrier Protein for Drugs
3.1. AFP—Whole Molecule (FL-AFP)
3.2. GIP-34 (Growth Inhibitor Peptide)
3.3. AFPep (Cyclic Version of GIP-8)
3.4. AFP-Inhibiting Fragments (AIFs)
4. RECAF (Alpha-Fetoprotein Receptor)
5. The Role of AFP in Cancer Immunotherapy
5.1. AFP and Immunotolerance in Hepatocellular Carcinoma
5.2. AFP as a Vaccine
5.3. TCR-T (T Cell Receptor–Transduced T Cells)
5.4. Dendritic Cell Vaccine
5.5. ICIs (Immune Checkpoint Inhibitors)
6. The Future of AFP in Oncology
6.1. Knockdown of the AFP Gene and AFP-siRNA
6.2. AFPmAb-PLGA-rhDCN
6.3. CAR-T (Chimeric Antigen Receptor T Cells)
6.4. The Combination of rhAFP with Taxanes and 1′-S-1′-Acetoxychavicol Acetate (ACA)
6.5. EGCG (Epigallocatechin-3-Gallate)
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACA | 1′-S-1′-acetoxychavicol acetate |
| AFP | alpha-fetoprotein |
| nAFP | native AFP |
| tAFP | tumor AFP |
| rhAFP | recombinant human AFP |
| AIFs | AFP-inhibiting fragments |
| AFPep | alpha-fetoprotein peptide |
| ATRA | all trans retinoic acid |
| APCs | antigen presenting cells |
| cAMP | cyclic adenosine monophosphate |
| cAMP-PKA | protein kinase A or cAMP-dependent protein kinase |
| c-kit | tyrosine-protein kinase KIT |
| CTL | cytotoxic T lymphocytes |
| CAR-T | chimeric antigen receptor T cells |
| CHOP | C/EBP homologous protein |
| CPP | cell-penetrating peptide |
| CXCR | CXC chemokine receptor |
| DC | dendritic cell |
| DDIT3 | DNA damage–inducible transcript |
| ECM | extracellular matrix |
| EpCAM | epithelial cell adhesion molecule |
| FAK | focal adhesion kinase |
| FL-AFP | full-length AFP |
| GADD153 | growth arrest- and DNA damage-inducible protein 153 |
| GIP | growth-inhibitory peptide |
| GPCR | G protein–coupled receptor |
| HCC | hepatocellular carcinoma |
| HRE | hormone response element |
| HLA | human leukocyte antigen |
| HSP | heat shock protein |
| ICISs | immune checkpoint inhibitors |
| IL | interleukin |
| IFN | interferon |
| K19 | keratin-19 |
| LPS | lipopolysaccharides |
| MHC | major histocompatibility complex |
| MDDCs | monocyte derived dendritic cells |
| MDSCs | myeloid-derived suppressor cells |
| G-MDSCs | granulocytic myeloid-derived suppressor cells |
| M-MDSCs | monocytic myeloid-derived suppressor cells |
| MDR | multiple drug resistance |
| MMP | matrix metalloproteinase |
| NK | natural killer |
| PD | programmed cell death protein |
| PD-L | programmed death ligand |
| PLGA | poly lactic-co-glycolic acid |
| PTEN | phosphatase and tensin homologue |
| RAR | retinoic acid receptor |
| RECAF | alpha-fetoprotein receptor |
| RISC | RNA-induced silencing complex |
| siRNA | small interfering RNA |
| SR | scavenger receptor |
| TCR-T | T cell receptor–transduced T cell |
| TCR | T cell receptor |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor alpha |
| TRAIL | tumor necrosis factor–related apoptosis-inducing ligand |
| Tregs | T regulatory cells |
References
- Mizejewski, G.J. Levels of alpha-fetoprotein during pregnancy and early infancy in normal and disease states. Obstet. Gynecol. Surv. 2003, 58, 804–826. [Google Scholar] [CrossRef] [PubMed]
- Mizejewski, G.J. Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp. Biol. Med. 2004, 229, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Krantz, D.A.; Hallahan, T.W.; Carmichael, J.B. Screening for Open Neural Tube Defects. Clin. Lab. Med. 2016, 36, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Glowska-Ciemny, J.; Pankiewicz, J.; Malewski, Z.; von Kaisenberg, C.; Kocylowski, R. Alpha-fetoprotein (AFP)—New aspects of a well-known marker in perinatology. Ginekol. Pol. 2022, 93, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Terentiev, A.A.; Moldogazieva, N.T. Alpha-fetoprotein: A renaissance. Tumour. Biol. 2013, 34, 2075–2091. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Liang, Y.; Si, R.; Jiang, Z.; Ma, B.; Gao, P. Knockdown of alpha-fetoprotein expression inhibits HepG2 cell growth and induces apoptosis. J. Cancer Res. Ther. 2018, 14, S634–S643. [Google Scholar] [CrossRef]
- Galle, P.R.; Foerster, F.; Kudo, M.; Chan, S.L.; Llovet, J.M.; Qin, S.; Schelman, W.R.; Chintharlapalli, S.; Abada, P.B.; Sherman, M.; et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 2019, 39, 2214–2229. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. Does alpha-fetoprotein contribute to the mortality and morbidity of human hepatocellular carcinoma? A commentary. J. Hepatocell. Carcinoma 2016, 3, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. Protein binding and interactions with alpha-fetoprotein (AFP): A review of multiple AFP cell surface receptors, intracytoplasmic binding, and inter-molecular complexing proteins. J. Mol. Cell Biol. Forecast 2019, 2, 1016. [Google Scholar]
- Lin, B.; Dong, X.; Wang, Q.; Li, W.; Zhu, M.; Li, M. AFP-Inhibiting Fragments for Drug Delivery: The Promise and Challenges of Targeting Therapeutics to Cancers. Front. Cell Dev. Biol. 2021, 9, 635476. [Google Scholar] [CrossRef]
- Munson, P.V.; Adamik, J.; Butterfield, L.H. Immunomodulatory impact of α-fetoprotein. Trends Immunol. 2022, 43, 438–448. [Google Scholar] [CrossRef]
- Mizejewski, G.J. Nonsecreted cytoplasmic alpha-fetoprotein: A newly discovered role in intracellular signaling and regulation. An update and commentary. Tumour. Biol. 2015, 36, 9857–9864. [Google Scholar] [CrossRef]
- Lin, B.; Zhu, M.; Wang, W.; Li, W.; Dong, X.; Chen, Y.; Lu, Y.; Guo, J.; Li, M. Structural basis for alpha fetoprotein-mediated inhibition of caspase-3 activity in hepatocellular carcinoma cells. Int. J. Cancer 2017, 141, 1413–1421. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, S.; Jiang, W.; Li, H.; Liu, Z.; Zhang, C.; McNutt, M.A.; Li, G. Impact of intracellular alpha fetoprotein on retinoic acid receptors-mediated expression of GADD153 in human hepatoma cell lines. Int. J. Cancer 2012, 130, 754–764. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Ding, H.; Guo, J.; et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018, 9, 1027. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhu, M.; Li, W.; Lin, B.; Dong, X.; Chen, Y.; Xie, X.; Guo, J.; Li, M. Alpha fetoprotein plays a critical role in promoting metastasis of hepatocellular carcinoma cells. J. Cell Mol. Med. 2016, 20, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. The alpha-fetoprotein (AFP) third domain: A search for AFP interaction sites of cell cycle proteins. Tumour. Biol. 2016, 37, 12697–12711. [Google Scholar] [CrossRef]
- Mizejewski, G.J. Alpha-fetoprotein structure and function: Relevance to isoforms, epitopes, and conformational variants. Exp. Biol. Med. 2001, 226, 377–408. [Google Scholar] [CrossRef]
- Mizejewski, G.J. The alpha-fetoprotein third domain receptor binding fragment: In search of scavenger and associated receptor targets. J. Drug Target. 2015, 23, 538–551. [Google Scholar] [CrossRef]
- Muehlemann, M.; Miller, K.D.; Dauphinee, M.; Mizejewski, G.J. Review of Growth Inhibitory Peptide as a biotherapeutic agent for tumor growth, adhesion, and metastasis. Cancer Metastasis Rev. 2005, 24, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.R.; Gildener-Leapman, N.; Narendran, A.; Lin, H.Y.; Lemanski, N.; Bennett, J.A.; Jacobson, H.I.; Andersen, T.T. Prevention of N-methyl-N-nitrosourea-induced breast cancer by alpha-fetoprotein (AFP)-derived peptide, a peptide derived from the active site of AFP. Clin. Cancer Res. 2005, 11, 8512–8520. [Google Scholar] [CrossRef] [Green Version]
- Tcherkassova, J.; Abramovich, C.; Moro, R.; Chen, C.; Schmit, R.; Gerber, A.; Moro, R. Combination of CA125 and RECAF biomarkers for early detection of ovarian cancer. Tumour. Biol. 2011, 32, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizejewski, G.J.; Mirowski, M.; Garnuszek, P.; Maurin, M.; Cohen, B.D.; Poiesz, B.J.; Posypanova, G.A.; Makarov, V.A.; Severin, E.S.; Severin, S.E. Targeted delivery of anti-cancer growth inhibitory peptides derived from human alpha-fetoprotein: Review of an International Multi-Center Collaborative Study. J. Drug Target. 2010, 18, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Mizejewski, G.J. Mechanism of Cancer Growth Suppression of Alpha-Fetoprotein Derived Growth Inhibitory Peptides (GIP): Comparison of GIP-34 versus GIP-8 (AFPep). Updates and Prospects. Cancers 2011, 3, 2709–2733. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. The alpha-fetoprotein-derived growth inhibitory peptide 8-mer fragment: Review of a novel anticancer agent. Cancer Biother. Radiopharm. 2007, 22, 73–98. [Google Scholar] [CrossRef]
- Jacobson, H.I.; Andersen, T.T.; Bennett, J.A. Development of an active site peptide analog of α-fetoprotein that prevents breast cancer. Cancer Prev. Res. (Phila) 2014, 7, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Joseph, L.C.; Bennett, J.A.; Kirschner, K.N.; Shields, G.C.; Hughes, J.; Lostritto, N.; Jacobson, H.I.; Andersen, T.T. Antiestrogenic and anticancer activities of peptides derived from the active site of alpha-fetoprotein. J. Pept. Sci. 2009, 15, 319–325. [Google Scholar] [CrossRef]
- Andersen, T.T.; Georgekutty, J.; Defreest, L.A.; Amaratunga, G.; Narendran, A.; Lemanski, N.; Jacobson, H.I.; Bennett, J.A. An alpha-fetoprotein-derived peptide reduces the uterine hyperplasia and increases the antitumour effect of tamoxifen. Br. J. Cancer 2007, 97, 327–333. [Google Scholar] [CrossRef]
- Bennett, J.A.; DeFreest, L.; Anaka, I.; Saadati, H.; Balulad, S.; Jacobson, H.I.; Andersen, T.T. AFPep: An anti-breast cancer peptide that is orally active. Breast Cancer Res. Treat. 2006, 98, 133–141. [Google Scholar] [CrossRef]
- Mansouri, W.; Fordyce, S.B.; Wu, M.; Jones, D.; Cohn, D.; Lin, Q.; Feustel, P.; Sharma, T.; Bennett, J.A.; Andersen, T.T. Efficacy and tolerability of AFPep, a cyclic peptide with anti-breast cancer properties. Toxicol. Appl. Pharmacol. 2018, 345, 10–18. [Google Scholar] [CrossRef]
- Mizejewski, G.J.; Muehlemann, M.; Dauphinee, M. Update of alpha fetoprotein growth-inhibitory peptides as biotherapeutic agents for tumor growth and metastasis. Chemotherapy 2006, 52, 83–90. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Ostroverkhova, D.S.; Kuzmich, N.N.; Kadochnikov, V.V.; Terentiev, A.A.; Porozov, Y.B. Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis. Int. J. Mol. Sci. 2020, 21, 893. [Google Scholar] [CrossRef] [Green Version]
- Zamorina, S.A.; Litvinova, L.S.; Yurova, K.A.; Khaziakhmatova, O.G.; Timganova, V.P.; Bochkova, M.S.; Khramtsov, P.V.; Raev, M.B.; Chereshnev, V.A. Role of α-Fetoprotein in Regulation of Proliferation and Functional Activity of Naïve T Cells and Immune Memory T Cells. Bull. Exp. Biol. Med. 2019, 167, 470–474. [Google Scholar] [CrossRef]
- Tower, A.M.; Trinward, A.; Lee, K.; Joseph, L.; Jacobson, H.I.; Bennett, J.A.; Andersen, T.T. AFPep, a novel drug for the prevention and treatment of breast cancer, does not disrupt the estrous cycle or fertility in rats. Oncol. Rep. 2009, 22, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Santos, P.M.; Menk, A.V.; Shi, J.; Tsung, A.; Delgoffe, G.M.; Butterfield, L.H. Tumor-Derived α-Fetoprotein Suppresses Fatty Acid Metabolism and Oxidative Phosphorylation in Dendritic Cells. Cancer Immunol. Res. 2019, 7, 1001–1012. [Google Scholar] [CrossRef]
- Ramadhani, D.; Maharani, R.; Gazzali, A.M.; Muchtaridi, M. Cyclic Peptides for the Treatment of Cancers: A Review. Molecules 2022, 27, 4428. [Google Scholar] [CrossRef]
- Tcherkassova, J.; Tsurkan, S.; Smirnova, G.; Borisova, J.; Moro, R.; Treshalina, H. Binding characterization of the targeting drug AIMPILA to AFP receptors in human tumor xenografts. Tumour. Biol. 2017, 39, 1010428317734815. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. Review of the putative cell-surface receptors for alpha-fetoprotein: Identification of a candidate receptor protein family. Tumour. Biol. 2011, 32, 241–258. [Google Scholar] [CrossRef]
- Grigor’eva, E.Y.; Treshchalina, E.M.; Lipengolts, A.A.; Smirnova, A.V.; Smirnova, G.B.; Borisova, Y.A.; Kalishyan, M.S. Biodistribution of Alpha-Fetoprotein-Containing Noncovalent Complex Aimpila with Antitumor Activity. Bull. Exp. Biol. Med. 2017, 164, 195–198. [Google Scholar] [CrossRef]
- Sedky, H.A.; Youssef, S.R.; Gamal, D.A.; Houssein, H.F.; Elsalakawy, W.A. First report of the unique expression of RECAF (receptor for alfa feto-protein) in adult B-NHL/CLL patients. Blood Res. 2020, 55, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Pak, V.N. α-fetoprotein-binding toxins and teratogens against cancer. Ther. Deliv. 2019, 10, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridder, D.A.; Weinmann, A.; Schindeldecker, M.; Urbansky, L.L.; Berndt, K.; Gerber, T.S.; Lang, H.; Lotz, J.; Lackner, K.J.; Roth, W.; et al. Comprehensive clinicopathologic study of alpha fetoprotein-expression in a large cohort of patients with hepatocellular carcinoma. Int. J. Cancer 2022, 150, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gong, H.; Liu, Q.; Wu, W.; Cheng, J.; Mei, Y.; Chen, Y.; Zheng, H.; Yu, X.; Zhong, S.; et al. Identification of an HLA-A*24:02-restricted α-fetoprotein signal peptide-derived antigen and its specific T-cell receptor for T-cell immunotherapy. Immunology 2020, 159, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Q. Alpha-Fetoprotein and Hepatocellular Carcinoma Immunity. Can. J. Gastroenterol. Hepatol. 2018, 2018, 9049252. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.S.; Zhang, C.; Chen, P.; Jin, S.J.; Jiang, G.Q. The prognostic correlation of AFP level at diagnosis with pathological grade, progression, and survival of patients with hepatocellular carcinoma. Sci. Rep. 2017, 7, 12870. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Semeniuk, D.J.; Jacobson, H.I.; Murgita, R.A. Similarity between natural and recombinant human alpha-fetoprotein as inhibitors of estrogen-dependent breast cancer growth. Breast Cancer Res. Treat. 1997, 45, 169–179. [Google Scholar] [CrossRef]
- Festin, S.M.; Bennett, J.A.; Fletcher, P.W.; Jacobson, H.I.; Shaye, D.D.; Andersen, T.T. The recombinant third domain of human alpha-fetoprotein retains the antiestrotrophic activity found in the full-length molecule. Biochim. Biophys. Acta 1999, 1427, 307–314. [Google Scholar] [CrossRef]
- Dudich, E. MM-093, a recombinant human alpha-fetoprotein for the potential treatment of rheumatoid arthritis and other autoimmune diseases. Curr. Opin. Mol. Ther. 2007, 9, 603–610. [Google Scholar]
- He, Y.; Hong, Y.; Mizejewski, G.J. Engineering α-fetoprotein-based gene vaccines to prevent and treat hepatocellular carcinoma: Review and future prospects. Immunotherapy 2014, 6, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Jiang, X.; Li, Q.; Liu, H.; Huang, L.; Wu, J.; Celis, E.; et al. Identification of α-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 2018, 68, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, V.N. Selective targeting of myeloid-derived suppressor cells in cancer patients through AFP-binding receptors. Future Sci. OA 2018, 5, FSO321. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.H.; Li, Y.G.; Liang, Z.W.; Chen, M.; Peng, M.L.; Tang, L.; Hu, H.D.; Ren, H. A DNA vaccine against chimeric AFP enhanced by HSP70 suppresses growth of hepatocellular carcinoma. Cancer Immunol. Immunother. 2007, 56, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Figley, C.; Long, R.; Park, D.; Carter, D.; Clements, V.K. Frontline Science: Myeloid-derived suppressor cells (MDSCs) facilitate maternal-fetal tolerance in mice. J. Leukoc. Biol. 2017, 101, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Baniyash, M. Myeloid-derived suppressor cells as intruders and targets: Clinical implications in cancer therapy. Cancer Immunol. Immunother. 2016, 65, 857–867. [Google Scholar] [CrossRef]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions with Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pho-Iam, T.; Punnakitikashem, P.; Somboonyosdech, C.; Sripinitchai, S.; Masaratana, P.; Sirivatanauksorn, V.; Sirivatanauksorn, Y.; Wongwan, C.; Nguyen, K.T.; Srisawat, C. PLGA nanoparticles containing α-fetoprotein siRNA induce apoptosis and enhance the cytotoxic effects of doxorubicin in human liver cancer cell line. Biochem. Biophys. Res. Commun. 2021, 53, 191–197. [Google Scholar] [CrossRef]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [Green Version]
- Chereshnev, V.A.; Timganova, V.P.; Zamorina, S.A.; Bochkova, M.S.; Khramtsov, P.V.; Kropaneva, M.D.; Raev, M.B. Role of α-fetoprotein in differentiation of regulatory T lymphocytes. Dokl. Biol. Sci. 2017, 477, 248–251. [Google Scholar] [CrossRef]
- Pardee, A.D.; Shi, J.; Butterfield, L.H. Tumor-derived α-fetoprotein impairs the differentiation and T cell stimulatory activity of human dendritic cells. J. Immunol. 2014, 193, 5723–5732. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Song, B.; Santos, P.M.; Butterfield, L.H. Hepatocellular cancer-derived alpha fetoprotein uptake reduces CD1 molecules on monocyte-derived dendritic cells. Cell Immunol. 2019, 335, 59–67. [Google Scholar] [CrossRef]
- Suryatenggara, J.; Wibowo, H.; Atmodjo, W.L.; Mathew, G. Characterization of alpha-fetoprotein effects on dendritic cell and its function as effector immune response activator. J. Hepatocell. Carcinoma 2017, 4, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Vujanovic, L.; Stahl, E.C.; Pardee, A.D.; Geller, D.A.; Tsung, A.; Watkins, S.C.; Gibson, G.A.; Storkus, W.J.; Butterfield, L.H. Tumor-Derived α-Fetoprotein Directly Drives Human Natural Killer-Cell Activation and Subsequent Cell Death. Cancer Immunol. Res. 2017, 5, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollaev, M.; Gorokhovets, N.; Nikolskaya, E.; Faustova, M.; Zabolotsky, A.; Sokol, M.; Tereshenko, O.; Zhunina, O.; Shvets, V.; Severin, E.; et al. Recombinant alpha-fetoprotein receptor-binding domain co-expression with polyglutamate tags facilitates in vivo folding in E. coli. Protein Expr. Purif. 2018, 143, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, N.N.; Abdolla, N.; Perfilyeva, Y.V.; Ostapchuk, Y.O.; Krasnoshtanov, V.K.; Kali, A.; Tleulieva, R. Daunorubicin conjugated with alpha-fetoprotein selectively eliminates myeloid-derived suppressor cells (MDSCs) and inhibits experimental tumor growth. Cancer Immunol. Immunother. 2018, 67, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Caraballo Galva, L.D.; Peng, Y.; Luo, X.; Zhu, W.; Yao, Y.; Ji, Y.; He, Y. Preclinical Studies of the Off-Target Reactivity of AFP158-Specific TCR Engineered T Cells. Front. Immunol. 2020, 11, 607. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Guo, H.; Jiang, R.; Lu, L.; Liu, T.; He, X. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive immu-notherapy in hepatocellular carcinoma. Tumour. Biol. 2016, 37, 799–806. [Google Scholar] [CrossRef]
- Docta, R.Y.; Ferronha, T.; Sanderson, J.P.; Weissensteiner, T.; Pope, G.R.; Bennett, A.D.; Pumphrey, N.J.; Ferjentsik, Z.; Quinn, L.L.; Wiedermann, G.E.; et al. Tuning T-Cell Receptor Affinity to Optimize Clinical Risk-Benefit When Targeting Alpha-Fetoprotein-Positive Liver Cancer. Hepatology 2019, 69, 2061–2075. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Cui, H.; Cai, L.; Zhu, W.; Yang, W.C.; Patrick, M.; Zhu, S.; Huang, J.; Yao, X.; Yao, Y.; et al. Selection of a Clinical Lead TCR Targeting Alpha-Fetoprotein-Positive Liver Cancer Based on a Balance of Risk and Benefit. Front. Immunol. 2020, 11, 623. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tatsumi, T.; Miyagi, T.; Tsunematsu, H.; Aketa, H.; Hosui, A.; Kanto, T.; Hiramatsu, N.; Hayashi, N.; Takehara, T. α-Fetoprotein impairs activation of natural killer cells by inhibiting the function of dendritic cells. Clin. Exp. Immunol. 2011, 165, 211–219. [Google Scholar] [CrossRef]
- Li, J.; Huang, S.; Zhou, Z.; Lin, W.; Chen, S.; Chen, M.; Ye, Y. Exosomes derived from rAAV/AFP-transfected dendritic cells elicit specific T cell-mediated immune responses against hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 4945–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, A.; Sadeghlar, F.; Ayub, T.H.; Schneider, C.; Möhring, C.; Zhou, T.; Mahn, R.; Bartels, A.; Praktiknjo, M.; Kornek, M.T.; et al. Alpha-Fetoprotein- and CD40Ligand-Expressing Dendritic Cells for Immunotherapy of Hepatocellular Carcinoma. Cancers 2021, 13, 3375. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, Y.; Lee, M.; Heo, M.K.; Song, J.S.; Kim, K.H.; Lee, H.; Yi, N.J.; Lee, K.W.; Suh, K.S.; et al. A phase I/IIa study of adjuvant immunotherapy with tumour antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Br. J. Cancer 2015, 113, 1666–1676. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Guo, H.; Jiang, R.; Lu, L.; Liu, T.; Zhang, Z.; He, X. Artificial antigen-presenting cells expressing AFP(158–166) peptide and interleukin-15 activate AFP-specific cytotoxic T lymphocytes. Oncotarget 2016, 7, 17579–17590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Kim, H.I.; Lim, J.; Shim, J.H. Role of the alpha-fetoprotein response in immune checkpoint inhibitor-based treatment of patients with hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2022, 148, 2069–2077. [Google Scholar] [CrossRef]
- Spahn, S.; Roessler, D.; Pompilia, R.; Gabernet, G.; Gladstone, B.P.; Horger, M.; Biskup, S.; Feldhahn, M.; Nahnsen, S.; Hilke, F.J.; et al. Clinical and Genetic Tumor Characteristics of Responding and Non-Responding Patients to PD-1 Inhibition in Hepatocellular Carcinoma. Cancers 2020, 12, 3830. [Google Scholar] [CrossRef]
- Hayashi, H.; Nakagawa, K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2020, 25, 818–830. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Liu, T.H.; Hsu, C.; Lu, L.C.; Shen, Y.C.; Lin, Z.Z.; Cheng, A.L.; Hsu, C.H. Early alpha-foetoprotein response associated with treatment efficacy of immune checkpoint inhibitors for advanced hepatocellular carcinoma. Liver. Int. 2019, 39, 2184–2189. [Google Scholar] [CrossRef]
- De Wilde, N.; Vonghia, L.; Francque, S.; De Somer, T.; Bagdadi, A.; Staub, E.; Lambrechts, J.; Bucalau, A.M.; Verset, G.; Van Steenkiste, C. Real-life multi-center retrospective analysis on nivolumab in difficult-to-treat patients with advanced hepatocellular carcinoma. World J. Hepatol. 2022, 14, 1608–1620. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, S.; Wang, Y.; Qu, Y.; Xue, J.; Mi, Y.; Wang, Y.; Luo, X.; Deng, Z.; Wang, G. Decorin-loaded poly lactic-co-glycolic acid nanoparticles modified by anti-alpha fetoprotein antibody: Preparation, proliferation inhibition and induced apoptosis effects on HepG2 cells in vitro. J. Pharm. Pharmacol. 2017, 69, 633–641. [Google Scholar] [CrossRef]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.M.; In, L.L.; Soh, T.L.; Azmi, M.N.; Ibrahim, H.; Awang, K.; Dudich, E.; Tatulov, E.; Nagoor, N.H. Recombinant human alpha fetoprotein synergistically potentiates the anti-cancer effects of 1’-S-1’-acetoxychavicol acetate when used as a complex against human tumours harbouring AFP-receptors. Oncotarget 2015, 6, 16151–16167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Liu, S.; Xu, J.; Li, W.; Duan, G.; Wang, H.; Yang, H.; Yang, Z.; Zhou, R. A new molecular mechanism underlying the EGCG-mediated autophagic modulation of AFP in HepG2 cells. Cell Death Dis. 2017, 8, e3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bimonte, S.; Albino, V.; Piccirillo, M.; Nasto, A.; Molino, C.; Palaia, R.; Cascella, M. Epigallocatechin-3-gallate in the prevention and treatment of hepatocellular carcinoma: Experimental findings and translational perspectives. Drug Des. Devel. Ther. 2019, 13, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Fan, X.; Bi, Y.; Zhou, Z.; Yuan, Y. Preparation of NIR-Responsive Gold Nanocages as Efficient Carrier for Controlling Release of EGCG in Anticancer Application. Front. Chem. 2022, 10, 926002. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Głowska-Ciemny, J.; Szymański, M.; Kuszerska, A.; Malewski, Z.; von Kaisenberg, C.; Kocyłowski, R. The Role of Alpha-Fetoprotein (AFP) in Contemporary Oncology: The Path from a Diagnostic Biomarker to an Anticancer Drug. Int. J. Mol. Sci. 2023, 24, 2539. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032539
Głowska-Ciemny J, Szymański M, Kuszerska A, Malewski Z, von Kaisenberg C, Kocyłowski R. The Role of Alpha-Fetoprotein (AFP) in Contemporary Oncology: The Path from a Diagnostic Biomarker to an Anticancer Drug. International Journal of Molecular Sciences. 2023; 24(3):2539. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032539
Chicago/Turabian StyleGłowska-Ciemny, Joanna, Marcin Szymański, Agata Kuszerska, Zbyszko Malewski, Constantin von Kaisenberg, and Rafał Kocyłowski. 2023. "The Role of Alpha-Fetoprotein (AFP) in Contemporary Oncology: The Path from a Diagnostic Biomarker to an Anticancer Drug" International Journal of Molecular Sciences 24, no. 3: 2539. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032539