
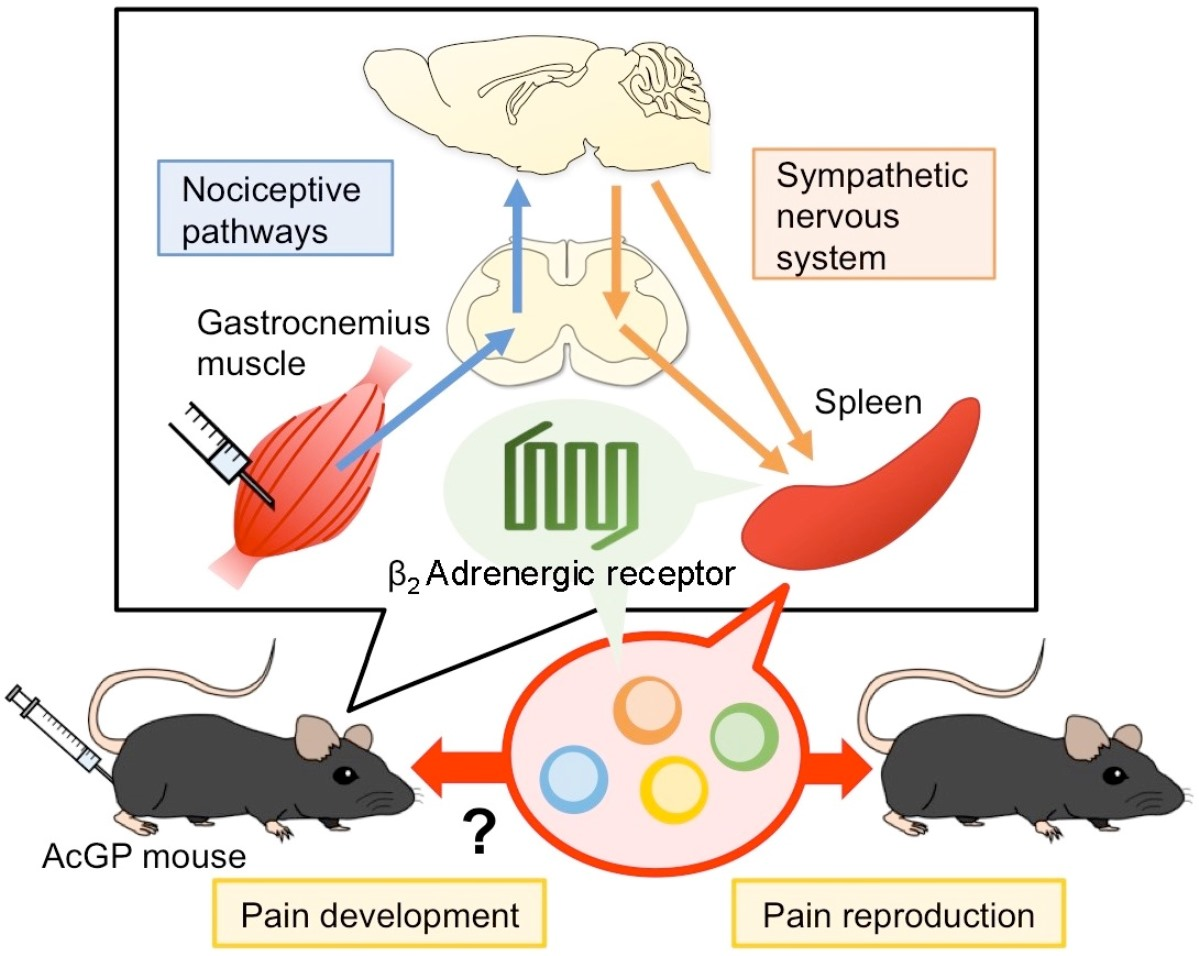
Peripheral Beta-2 Adrenergic Receptors Mediate the Sympathetic Efferent Activation from Central Nervous System to Splenocytes in a Mouse Model of Fibromyalgia
 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
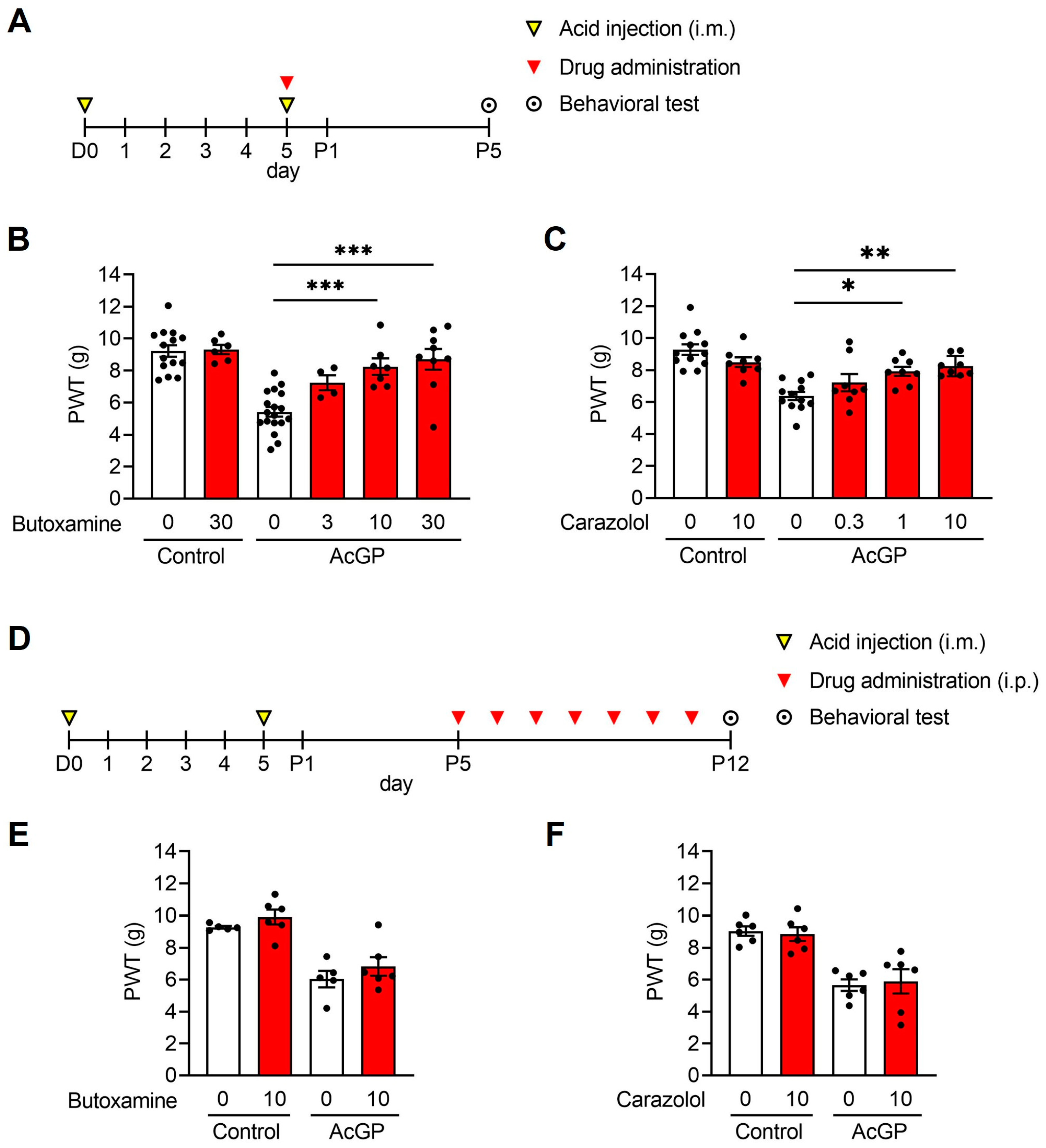
2.1. β2-Adrenergic Receptors Are Involved in the Development but Not in the Maintenance of the Acid-Induced Mechanical Hypersensitivity
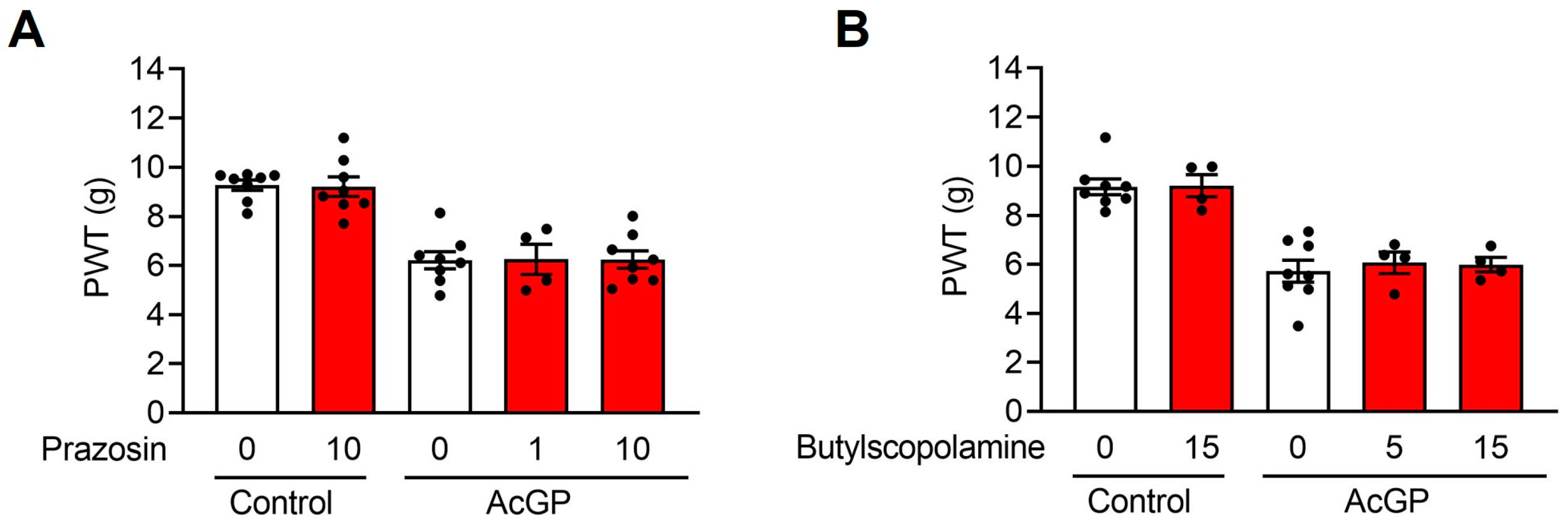
2.2. Neither α1-Adrenergic nor Muscarinic Receptors Contribute to the Acid-Induced Mechanical Hypersensitivity
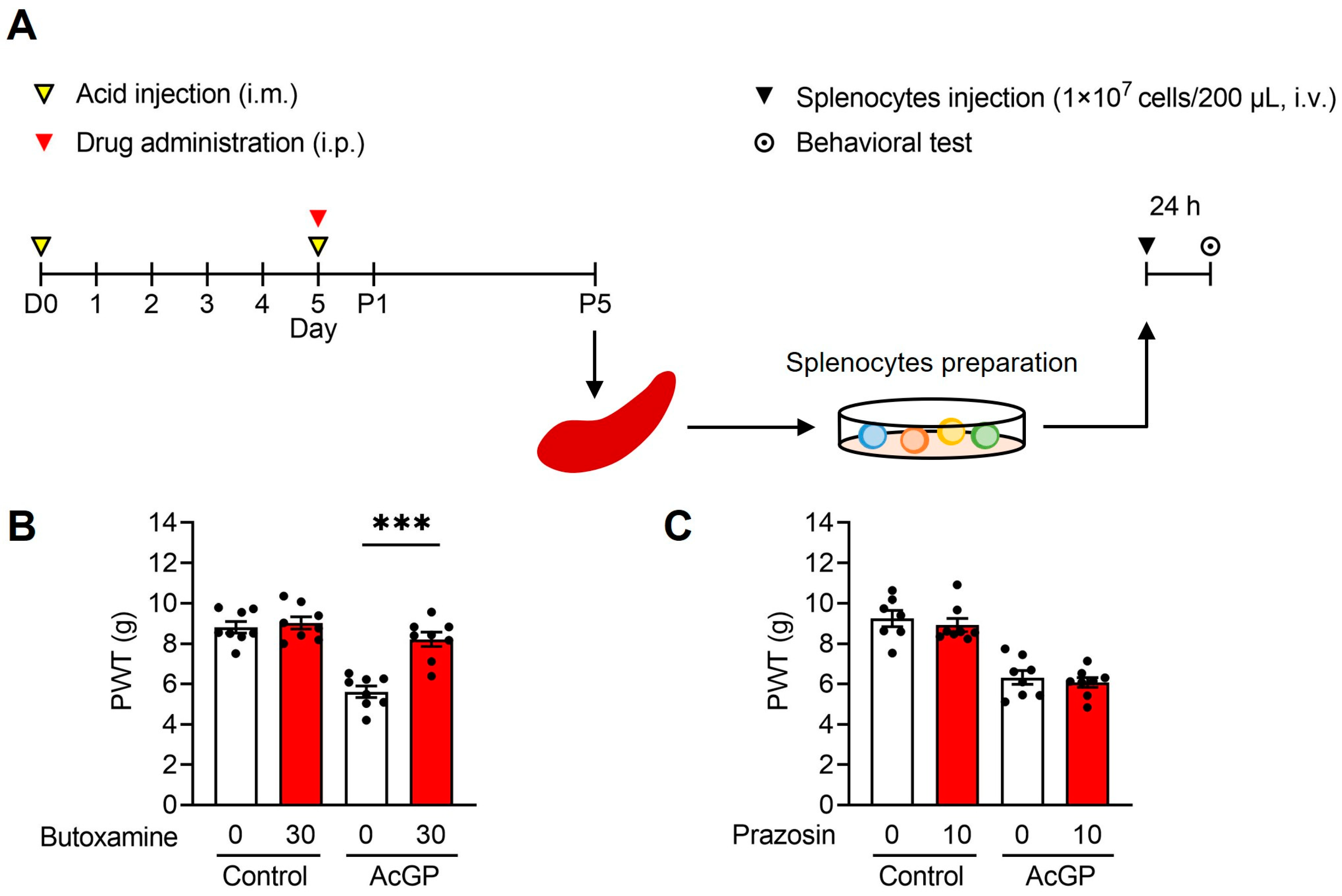
2.3. Adoptive Transfer of Splenocytes from AcGP Mice Stimulated by β2-Adrenergic Receptor Activation Reproduces the Mechanical Hypersensitivity in Naïve Recipient Mice
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. AcGP Mouse Model
4.3. Drug Treatments
4.4. Behavioral Test
4.5. Isolation and Adoptive Transfer of Splenocytes
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siracusa, R.; Paola, R.D.; Cuzzocrea, S.; Impellizzeri, D. Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. Int. J. Mol. Sci. 2021, 22, 3891. [Google Scholar] [CrossRef] [PubMed]
- Häuser, W.; Ablin, J.; Fitzcharles, M.A.; Littlejohn, G.; Luciano, J.V.; Usui, C.; Walitt, B. Fibromyalgia. Nat. Rev. Dis. Primers 2015, 1, 15022. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.B.; Lange, G.; Ciccone, D.S.; Liu, W.C.; Steffener, J.; Natelson, B.H. Functional imaging of pain in patients with primary fibromyalgia. J. Rheumatol. 2004, 31, 364–378. [Google Scholar]
- Ji, R.R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Höglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Serra, J.; Collado, A.; Solà, R.; Antonelli, F.; Torres, X.; Salgueiro, M.; Quiles, C.; Bostock, H. Hyperexcitable C nociceptors in fibromyalgia. Ann. Neurol. 2014, 75, 196–208. [Google Scholar] [CrossRef]
- Teng, H.W.; Tani, J.; Chang, T.S.; Chen, H.J.; Lin, Y.C.; Lin, C.S.; Sung, J.Y. Altered sensory nerve excitability in fibromyalgia. J. Formos Med. Assoc. 2021, 120, 1611–1619. [Google Scholar]
- Buskila, D.; Sarzi-Puttini, P. Fibromyalgia and autoimmune diseases: The pain behind autoimmunity. Isr. Med. Assoc. J. 2008, 10, 77–78. [Google Scholar]
- Bote, M.E.; García, J.J.; Hinchado, M.D.; Ortega, E. Inflammatory/stress feedback dysregulation in women with fibromyalgia. Neuroimmunomodulation 2012, 19, 343–351. [Google Scholar]
- Ang, D.C.; Moore, M.N.; Hilligoss, J.; Tabbey, R. MCP-1 and IL-8 as pain biomarkers in fibromyalgia: A pilot study. Pain Med. 2011, 12, 1154–1161. [Google Scholar] [PubMed]
- Qiu, Y.; Zhang, T.J.; Meng, L.B.; Cheng, X.T.; Hua, Z. Bioinformatics analysis of gene and microRNA targets for fibromyalgia. Clin. Exp. Rheumatol. 2021, 39, 21–31. [Google Scholar]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy. J. Clin. Med. 2020, 9, 1814. [Google Scholar]
- Sluka, K.A.; Kalra, A.; Moore, S.A. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve 2001, 24, 37–46. [Google Scholar] [CrossRef]
- Khasar, S.G.; Miao, F.J.; Jänig, W.; Levine, J.D. Vagotomy-induced enhancement of mechanical hyperalgesia in the rat is sympathoadrenal-mediated. J. Neurosci. 1998, 18, 3043–3049. [Google Scholar]
- Khasar, S.G.; Green, P.G.; Levine, J.D. Repeated sound stress enhances inflammatory pain in the rat. Pain 2005, 116, 79–86. [Google Scholar] [PubMed]
- Nishiyori, M.; Ueda, H. Prolonged gabapentin analgesia in an experimental mouse model of fibromyalgia. Mol. Pain 2008, 64, 52. [Google Scholar]
- Ueda, H.; Neyama, H. LPA1 receptor involvement in fibromyalgia-like pain induced by intermittent psychological stress, empathy. Neurobiol. Pain 2017, 1, 16–25. [Google Scholar]
- Nagakura, Y.; Oe, T.; Aoki, T.; Matsuoka, N. Biogenic amine depletion causes chronic muscular pain and tactile allodynia accompanied by depression: A putative animal model of fibromyalgia. Pain 2009, 146, 26–33. [Google Scholar]
- Ueda, H.; Dozono, N.; Tanaka, K.; Kaneko, S.; Neyama, H.; Uchida, H. Allodynia by Splenocytes From Mice With Acid-Induced Fibromyalgia-Like Generalized Pain and Its Sexual Dimorphic Regulation by Brain Microglia. Front. Neurosci. 2020, 14, 600166. [Google Scholar] [CrossRef]
- Yokoyama, T.; Maeda, Y.; Audette, K.M.; Sluka, K.A. Pregabalin reduces muscle and cutaneous hyperalgesia in two models of chronic muscle pain in rats. J. Pain 2007, 8, 422–429. [Google Scholar]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [PubMed] [Green Version]
- Bellinger, D.L.; Felten, S.Y.; Lorton, D.; Felten, D.L. Origin of noradrenergic innervation of the spleen in rats. Brain Behav. Immun. 1989, 3, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Peterson, R.G.; Shea, P.A.; Schmedtje, J.F.; Bauer, D.C.; Felten, D.L. Sympathetic innervation of murine thymus and spleen: Evidence for a functional link between the nervous and immune systems. Brain Res. Bull. 1981, 6, 83–94. [Google Scholar] [CrossRef]
- Davies, B.N.; Withrington, P.G. The actions of drugs on the smooth muscle of the capsule and blood vessels of the spleen. Pharmacol. Rev. 1973, 25, 373–413. [Google Scholar] [PubMed]
- Madden, K.S.; Sanders, V.M.; Felten, D.L. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 417–448. [Google Scholar] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve--an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Maletic, V.; Raison, C.L. Neurobiology of depression, fibromyalgia and neuropathic pain. Front. Biosci. (Landmark Ed.) 2009, 14, 5291–5338. [Google Scholar]
- Fernández-López, A.; Revilla, V.; Candelas, M.A.; Aller, M.I.; Soria, C.; Pazos, A. Identification of beta-adrenoceptors in rat lymph nodes and spleen: An autoradiographic study. Eur. J. Pharmacol. 1994, 262, 283–286. [Google Scholar]
- Kohm, A.P.; Sanders, V.M. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol. Rev. 2001, 53, 487–525. [Google Scholar]
- Elsinga, P.H.; Hendrikse, N.H.; Bart, J.; van Waarde, A.; Vaalburg, W. Positron emission tomography studies on binding of central nervous system drugs and P-glycoprotein function in the rodent brain. Mol. Imaging Biol. 2005, 7, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Khasar, S.G.; McCarter, G.; Levine, J.D. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J. Neurophysiol. 1999, 81, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D. Autonomic regulation of cellular immune function. Auton. Neurosci. 2014, 182, 15–41. [Google Scholar] [PubMed]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [PubMed]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol. 2016, 594, 5781–5790. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar]
- Todorov, L.D.; Mihaylova-Todorova, S.T.; Bjur, R.A.; Westfall, D.P. Differential cotransmission in sympathetic nerves: Role of frequency of stimulation and prejunctional autoreceptors. J. Pharmacol. Exp. Ther. 1999, 290, 241–246. [Google Scholar]
- Guggino, G.; Schinocca, C.; Lo Pizzo, M.; Di Liberto, D.; Garbo, D.; Raimondo, S.; Alessandro, R.; Brighina, F.; Ruscitti, P.; Giacomelli, R.; et al. T helper 1 response is correlated with widespread pain, fatigue, sleeping disorders and the quality of life in patients with fibromyalgia and is modulated by hyperbaric oxygen therapy. Clin. Exp. Rheumatol. 2020, 38, 1275. [Google Scholar]
- Sharif, K.; Watad, A.; Bragazzi, N.L.; Lichtbroun, M.; Amital, H.; Shoenfeld, Y. Physical activity and autoimmune diseases: Get moving and manage the disease. Autoimmun. Rev. 2018, 17, 53–72. [Google Scholar] [CrossRef]
- Liu, Y.; Rui, X.X.; Shi, H.; Qiu, Y.H.; Peng, Y.P. Norepinephrine Inhibits Th17 Cells via β2-Adrenergic Receptor (β2-AR) Signaling in a Mouse Model of Rheumatoid Arthritis. Med. Sci. Monit. 2018, 24, 1196–1204. [Google Scholar] [CrossRef]
- Carvajal Gonczi, C.M.; Tabatabaei Shafiei, M.; East, A.; Martire, E.; Maurice-Ventouris, M.H.I.; Darlington, P.J. Reciprocal modulation of helper Th1 and Th17 cells by the β2-adrenergic receptor agonist drug terbutaline. FEBS J. 2017, 284, 3018–3028. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.J.; Chen, C.C.; Yang, H.W.; Chang, Y.T.; Bai, S.W.; Chen, C.C.; Yen, C.T.; Min, M.Y. Role of extracellular signal-regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid-induced muscle pain mice. J. Neurosci. 2011, 31, 2258–2270. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.K.; Liu, I.Y.; Chang, Y.T.; Chen, Y.C.; Chen, C.C.; Yen, C.T.; Shin, H.S.; Chen, C.C. Ca(v)3.2 T-type Ca2+ channel-dependent activation of ERK in paraventricular thalamus modulates acid-induced chronic muscle pain. J. Neurosci. 2010, 30, 10360–10368. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.N.; Lee, C.H.; Lin, S.H.; Wong, C.W.; Sun, W.H.; Wood, J.N.; Chen, C.C. Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol. Pain 2014, 10, 40. [Google Scholar] [PubMed]
- Zhang, F.F.; Morioka, N.; Abe, H.; Fujii, S.; Miyauchi, K.; Nakamura, Y.; Hisaoka-Nakashima, K.; Nakata, Y. Stimulation of spinal dorsal horn β2-adrenergic receptor ameliorates neuropathic mechanical hypersensitivity through a reduction of phosphorylation of microglial p38 MAP kinase and astrocytic c-jun N-terminal kinase. Neurochem. Int. 2016, 101, 144–155. [Google Scholar] [PubMed]
- Chen, N.; Ge, M.M.; Li, D.Y.; Wang, X.M.; Liu, D.Q.; Ye, D.W.; Tian, Y.K.; Zhou, Y.Q.; Chen, J.P. β2-adrenoreceptor agonist ameliorates mechanical allodynia in paclitaxel-induced neuropathic pain via induction of mitochondrial biogenesis. Biomed. Pharmacother. 2021, 144, 112331. [Google Scholar] [CrossRef]
- Yalcin, I.; Tessier, L.H.; Petit-Demoulière, N.; Waltisperger, E.; Hein, L.; Freund-Mercier, M.J.; Barrot, M. Chronic treatment with agonists of beta(2)-adrenergic receptors in neuropathic pain. Exp. Neurol. 2010, 221, 115–121. [Google Scholar]
- Grace, P.M.; Hutchinson, M.R.; Bishop, A.; Somogyi, A.A.; Mayrhofer, G.; Rolan, P.E. Adoptive transfer of peripheral immune cells potentiates allodynia in a graded chronic constriction injury model of neuropathic pain. Brain Behav. Immun. 2011, 25, 503–513. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. BMC Vet. Res. 2020, 16, 242. [Google Scholar] [CrossRef]
- Yen, C.M.; Hsieh, C.L.; Lin, Y.W. Electroacupuncture reduces chronic fibromyalgia pain through attenuation of transient receptor potential vanilloid 1 signaling pathway in mouse brains. Iran J. Basic Med. Sci. 2020, 23, 894–900. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figure | Group | Total Number of Animals Used | Sample Size | Number of Animals Excluded 1 | |
|---|---|---|---|---|---|
| Figure 1B | Butoxamine concentration (mg/kg) | ||||
| Control | 0 | 16 | 14 | 2 | |
| 30 | 6 | 6 | 0 | ||
| AcGP | 0 | 18 | 18 | 0 | |
| 3 | 4 | 4 | 0 | ||
| 10 | 8 | 7 | 1 | ||
| 30 | 9 | 9 | 0 | ||
| Figure 1C | Carazolol concentration (mg/kg) | ||||
| Control | 0 | 12 | 12 | 0 | |
| 10 | 8 | 8 | 0 | ||
| AcGP | 0 | 12 | 12 | 0 | |
| 0.3 | 8 | 8 | 0 | ||
| 1 | 8 | 8 | 0 | ||
| 10 | 8 | 8 | 0 | ||
| Figure 1E | Butoxamine concentration (mg/kg) | ||||
| Control | 0 | 5 | 5 | 0 | |
| 30 | 6 | 6 | 0 | ||
| AcGP | 0 | 5 | 5 | 0 | |
| 30 | 6 | 6 | 0 | ||
| Figure 1F | Carazolol concentration (mg/kg) | ||||
| Control | 0 | 6 | 6 | 0 | |
| 10 | 7 | 6 | 1 | ||
| AcGP | 0 | 6 | 6 | 0 | |
| 10 | 6 | 6 | 0 | ||
| Figure 2A | Prazosin concentration (mg/kg) | ||||
| Control | 0 | 8 | 8 | 0 | |
| 10 | 8 | 8 | 0 | ||
| AcGP | 0 | 8 | 8 | 0 | |
| 1 | 4 | 4 | 0 | ||
| 10 | 8 | 8 | 0 | ||
| Figure 2B | Butylscopolamine concentration (mg/kg) | ||||
| Control | 0 | 8 | 8 | 0 | |
| 15 | 4 | 4 | 0 | ||
| AcGP | 0 | 8 | 8 | 0 | |
| 5 | 4 | 4 | 0 | ||
| 15 | 4 | 4 | 0 | ||
| Figure 3B | Butoxamine concentration (mg/kg) | ||||
| Control | 0 | 8 | 8 | 0 | |
| 30 | 8 | 8 | 0 | ||
| AcGP | 0 | 8 | 8 | 0 | |
| 30 | 9 | 8 | 1 | ||
| Figure 3C | Prazosin concentration (mg/kg) | ||||
| Control | 0 | 7 | 7 | 0 | |
| 10 | 8 | 8 | 0 | ||
| AcGP | 0 | 8 | 8 | 0 | |
| 10 | 8 | 8 | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, S.; Dozono, N.; Tobori, S.; Nagayasu, K.; Kaneko, S.; Shirakawa, H.; Ueda, H. Peripheral Beta-2 Adrenergic Receptors Mediate the Sympathetic Efferent Activation from Central Nervous System to Splenocytes in a Mouse Model of Fibromyalgia. Int. J. Mol. Sci. 2023, 24, 3465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043465
Yamashita S, Dozono N, Tobori S, Nagayasu K, Kaneko S, Shirakawa H, Ueda H. Peripheral Beta-2 Adrenergic Receptors Mediate the Sympathetic Efferent Activation from Central Nervous System to Splenocytes in a Mouse Model of Fibromyalgia. International Journal of Molecular Sciences. 2023; 24(4):3465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043465
Chicago/Turabian StyleYamashita, Shiori, Naoki Dozono, Shota Tobori, Kazuki Nagayasu, Shuji Kaneko, Hisashi Shirakawa, and Hiroshi Ueda. 2023. "Peripheral Beta-2 Adrenergic Receptors Mediate the Sympathetic Efferent Activation from Central Nervous System to Splenocytes in a Mouse Model of Fibromyalgia" International Journal of Molecular Sciences 24, no. 4: 3465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043465