
Pediatric-Onset Epilepsy and Developmental Epileptic Encephalopathies Followed by Early-Onset Parkinsonism

Abstract
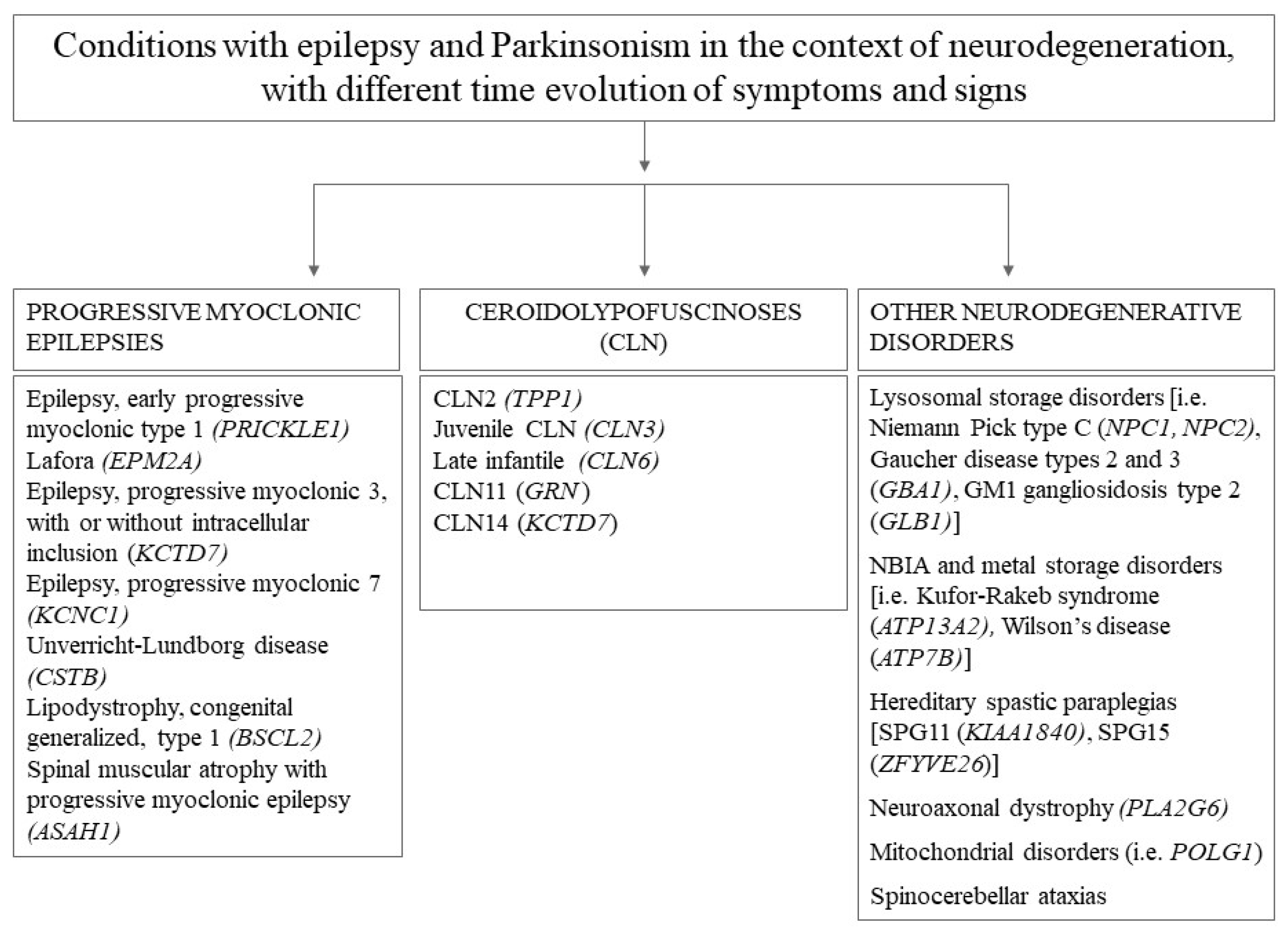
:1. Introduction
2. Materials and Methods
3. Results
3.1. Neurodevelopmental Disorders and DE-EE (Table 1)
3.1.1. PPP2R5D (Protein Phosphatase 2 Regulatory Subunit B’Delta)
3.1.2. FOXG1 (Forkhead Box G1)
3.1.3. STXBP1 (Syntaxin-Binding Protein 1)
3.1.4. Rett Syndrome Secondary to methyl CpG Binding Protein 2 (MECP2) Variants
3.1.5. Dravet Syndrome Due to Sodium Voltage-Gated Channel Alpha Subunit 1 (SCN1A) Variants
3.1.6. TBC1 Domain Family Member 24 (TBC1D24)
3.1.7. FRRS1L

{kind=link}
{kind=link}
{kind=link}
| Gene/Condition Name | Inheritance | Cognition | Additional NDD Features | Neurological Examination and MDs | Syndromic Features | Epilepsy | Parkinsonism, Onset Age |
|---|---|---|---|---|---|---|---|
| PPP2R5D | AD | DD, ID | ASD, speech impairment | Infantile hypotonia, progressive hypertonia | Macrocephaly, facial dysmorphism, variable skeletal, endocrine, and cardiac defects | Possible | 20–40 y |
| FOXG1 | AD | DD, severe cognitive impairment | Absent language, autistic features | Hyperkinetic MDs | severe postnatal microcephaly | DE | Teenage years/adulthood |
| STXBP1 | AD | Severe DD, ID | ASD | Hyperkinetic MD | / | DE-EE | Teenage years |
| Rett Syndrome MECP2 | XL | Absent/unremarkable before regression phase | Regression of motor and language function | Hypotonia, hyperkinetic MD | Acquired microcephaly, progressive scoliosis | Non-specific epilepsy or DE-EE | Teenage years/adulthood |
| Dravet Syndrome (SCN1A) | AD | Variable DD/ID | Possible autistic features | Initially normal, then ataxia, spasticity, dysarthria, crouch gait | Clinical criteria for Rett syndrome diagnosis | DE-EE | Adulthood |
| TBC1D24 | AD | DD, ID | / | Chronic and paroxysmal hyperkinetic MD | Possible cerebro-cerebellar malformation | Usually drug-resistant | Early 20′s |
| FRRS1L | AD | Normal early development followed by regression with GDD | Additional NDD features | Diffuse hypotonia, chorea, pyramidal signs | / | Multiple seizure types, mainly clonic/myoclonic | Late adolescence |
3.1.8. Syndromic Conditions with CNVS
22q11.2 Deletion Syndrome
3.1.9. 6q26-q27 Deletion
3.1.10. Chromosomopathies
3.2. Neurodegeneration
3.2.1. WD Repeat Domain 45 (WDR45)
3.2.2. IT15
3.3. Juvenile Parkinsonism Genes
3.3.1. KCND3
3.3.2. ATP6AP2
3.3.3. DNAJC6
3.3.4. SYNJ1
3.3.5. NR4A2
3.3.6. RAB39B
3.3.7. MECP2 Pathogenic Variant p.(Ala140Val) in Males
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Niemann, N.; Jankovic, J. Juvenile Parkinsonism: Differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Rocca, W.A. Incidence and distribution of Parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology 1999, 52, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Neuromelanin and Parkinson’s disease. J. Neural Transm. Suppl. 1983, 19, 121–141. [Google Scholar] [PubMed]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Zhang, M.; Mu, H.; Shang, Z.; Kang, K.; Lv, H.; Duan, L.; Li, J.; Chen, X.; Teng, Y.; Jiang, Y.; et al. Genome-wide pathway-based association analysis identifies risk pathways associated with Parkinson’s disease. Neuroscience 2017, 340, 398–410. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Specchio, N.; Curatolo, P. Developmental and epileptic encephalopathies: What we do and do not know. Brain 2021, 144, 32–43. [Google Scholar] [CrossRef]
- Papandreou, A.; Danti, F.R.; Spaull, R.; Leuzzi, V.; Mctague, A.; Kurian, M.A. The expanding spectrum of movement disorders in genetic epilepsies. Dev. Med. Child Neurol. 2020, 62, 178–191. [Google Scholar] [CrossRef]
- Morales-Briceño, H.; Mohammad, S.S.; Post, B.; Fois, A.F.; Dale, R.C.; Tchan, M.; Fung, V.S.C. Clinical and neuroimaging phenotypes of genetic Parkinsonism from infancy to adolescence. Brain 2020, 143, 751–770. [Google Scholar] [CrossRef]
- Leuzzi, V.; Nardecchia, F.; Pons, R.; Galosi, S. Parkinsonism in children: Clinical classification and etiological spectrum. Parkinsonism. Relat. Disord. 2021, 82, 150–157. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Rivers, A.M.; Audlin, S.; Virshup, D.M. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 1996, 271, 22081–22089. [Google Scholar] [CrossRef]
- Hetzelt, K.L.M.L.; Kerling, F.; Kraus, C.; Rauch, C.; Thiel, C.T.; Winterholler, M.; Reis, A.; Zweier, C. Early-onset Parkinsonism in PPP2R5D-related neurodevelopmental disorder. Eur. J. Med. Genet. 2021, 64, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Wirth, T.; Hubsch, C.; Németh, A.H.; Okur, V.; Anheim, M.; Drouot, N.; Tranchant, C.; Rudolf, G.; Chelly, J.; et al. Early-Onset Parkinsonism Is a Manifestation of the PPP2R5D p.E200K Mutation. Ann. Neurol. 2020, 88, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with Parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunctionand oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Florian, C.; Bahi-Buisson, N.; Bienvenu, T. FOXG1-Related Disorders: From Clinical Description to Molecular Genetics. Mol. Syndromol. 2012, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.C.; Singh, S.; Wang, H.P.; Hsu, C.J.; Hu, S.C.; Lee, W.T. FOXG1-Related Syndrome: From Clinical to Molecular Genetics and Pathogenic Mechanisms. Int. J. Mol. Sci. 2019, 20, 4176. [Google Scholar] [CrossRef]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Bardai, F.H.; Ma, C.; Price, V.; Rawat, V.; Verma, P.; Narayanan, V.; D’Mello, S.R. Isoform-specific toxicity of Mecp2 in postmitotic neurons: Suppression of neurotoxicity by FoxG1. J. Neurosci. 2012, 32, 2846–2855. [Google Scholar] [CrossRef]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, A.; Schneider, R.B.; Augustine, E.F.; Ng, J.; Mankad, K.; Meyer, E.; McTague, A.; Ngoh, A.; Hemingway, C.; Robinson, R.; et al. Delineation of the movement disorders associated with FOXG1 mutations. Neurology 2016, 86, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Méneret, A.; Mignot, C.; An, I.; Habert, M.O.; Jacquette, A.; Vidailhet, M.; Bienvenu, T.; Roze, E. Generalized dystonia, athetosis, and Parkinsonism associated with FOXG1 mutation. Mov. Disord. 2012, 27, 160–161. [Google Scholar] [CrossRef]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef]
- Di Meglio, C.; Lesca, G.; Villeneuve, N.; Lacoste, C.; Abidi, A.; Cacciagli, P.; Altuzarra, C.; Roubertie, A.; Afenjar, A.; Renaldo-Robin, F.; et al. Epileptic patients with de novo STXBP1 mutations: Key clinical features based on 24 cases. Epilepsia 2015, 56, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Khaikin, Y.; Mercimek-Mahmutoglu, S. STXBP1 encephalopathy with epilepsy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Álvarez Bravo, G.; Yusta Izquierdo, A. The adult motor phenotype of Dravet syndrome is associated with mutation of the STXBP1 gene and responds well to cannabidiol treatment. Seizure 2018, 60, 68–70. [Google Scholar] [CrossRef]
- Keogh, M.J.; Daud, D.; Pyle, A.; Duff, J.; Griffin, H.; He, L.; Alston, C.L.; Steele, H.; Taggart, S.; Basu, A.P.; et al. A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late- onset juvenile-onset Parkinsonism. Neurogenetics 2015, 16, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Crosiers, D.; Balagura, G.; Bonardi, C.M.; Basu, A.; Cantalupo, G.; Chiesa, V.; Christensen, J.; Dalla Bernardina, B.; Ellis, C.A.; et al. Natural History Study of STXBP1-Developmental and Epileptic Encephalopathy Into Adulthood. Neurology 2022, 99, e221–e233. [Google Scholar] [CrossRef]
- Lanoue, V.; Chai, Y.J.; Brouillet, J.Z.; Weckhuysen, S.; Palmer, E.E.; Collins, B.M.; Meunier, F.A. STXBP1 encephalopathy: Connecting neurodevelopmental disorders with α-synucleinopathies? Neurology 2019, 93, 114–123. [Google Scholar] [CrossRef]
- Spagnoli, C.; Fusco, C.; Pisani, F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes 2021, 12, 1157. [Google Scholar] [CrossRef]
- Temudo, T.; Ramos, E.; Dias, K.; Barbot, C.; Vieira, J.P.; Moreira, A.; Calado, E.; Carrilho, I.; Oliveira, G.; Levy, A.; et al. Movement disorders in Rett syndrome: An analysis of 60 patients with detected MECP2 mutation and correlation with mutation type. Mov. Disord. 2008, 23, 1384–1390. [Google Scholar] [CrossRef]
- Humphreys, P.; Barrowman, N. The Incidence and Evolution of Parkinsonian Rigidity in Rett Syndrome: A Pilot Study. Can. J. Neurol. Sci. 2016, 43, 567–573. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, P.M.; Jankovic, J.; Glaze, D.G.; Schultz, R.; Percy, A.K. Extrapyramidal involvement in Rett’s syndrome. Neurology 1990, 40, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.R.; Downs, J.; Bebbington, A.; Jacoby, P.; Girdler, S.; Kaufmann, W.E.; Leonard, H. Change in gross motor abilities of girls and women with rett syndrome over a 3- to 4-year period. J. Child Neurol. 2011, 26, 1237–1245. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology 1995, 45, 1581–1586. [Google Scholar] [CrossRef]
- Armstrong, D.D. Neuropathology of Rett syndrome. J. Child Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Percy, A.K.; Glaze, D.G.; Butler, I.J.; Riccardi, V.M. Reduction of biogenic amine levels in the Rett syndrome. N. Engl. J. Med. 1985, 313, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Milstien, S.; Butler, I.J.; Smith, E.O.; Kaufman, S.; Glaze, D.G.; Percy, A.K. Cerebrospinal fluid biogenic amines and biopterin in Rett syndrome. Ann. Neurol. 1989, 25, 56–60. [Google Scholar] [CrossRef]
- Perry, T.L.; Dunn, H.G.; Ho, H.H.; Crichton, J.U. Cerebrospinal fluid values for monoamine metabolites, gamma-aminobutyric acid, and other amino compounds in Rett syndrome. J. Pediatr. 1988, 112, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Lekman, A.; Witt-Engerström, I.; Holmberg, B.; Percy, A.; Svennerholm, L.; Hagberg, B. CSF and urine biogenic amine metabolites in Rett syndrome. Clin. Genet. 1990, 37, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; Mandel-Brehm, C.; Chao, H.T.; Ward, C.S.; Fyffe-Maricich, S.L.; Ren, J.; Hyland, K.; Thaller, C.; Maricich, S.M.; Humphreys, P.; et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2009, 106, 21966–21971. [Google Scholar] [CrossRef]
- Rodda, J.M.; Scheffer, I.E.; McMahon, J.M.; Berkovic, S.F.; Graham, H.K. Progressive gait deterioration in adolescents with Dravet syndrome. Arch. Neurol. 2012, 69, 873e8. [Google Scholar] [CrossRef] [PubMed]
- Gitiaux, C.; Chemaly, N.; Quijano-Roy, S.; Barnerias, C.; Desguerre, I.; Hully, M.; Chiron, C.; Dulac, O.; Nabbout, R. Motor neuropathy contributes to crouching in patients with Dravet syndrome. Neurology 2016, 87, 277–281. [Google Scholar] [CrossRef]
- Fasano, A.; Borlot, F.; Lang, A.; Andrade, D. Antecollis and levodopa-responsive Parkinsonism are late features of Dravet syndrome. Neurology 2014, 82, 2250–2251. [Google Scholar] [CrossRef]
- Aljaafari, D.; Fasano, A.; Nascimento, F.A.; Lang, A.E.; Andrade, D.M. Adult motor phenotype differentiates Dravet syndrome from Lennox-Gastaut syndrome and links SCN1A to early onset parkinsonian features. Epilepsia 2017, 58, e44e8. [Google Scholar] [CrossRef]
- Wyers, L.; Van de Walle, P.; Hoornweg, A.; Tepes Bobescu, I.; Verheyen, K.; Ceulemans, B.; Schoonjans, A.; Desloovere, K.; Hallemans, A. Gait deviations in patients with Dravet syndrome: A systematic review. Eur. J. Paediatr. Neurol. 2019, 23, 357–367. [Google Scholar] [CrossRef]
- Lukk, M.; Kapushesky, M.; Nikkilä, J.; Parkinson, H.; Goncalves, A.; Huber, W.; Ukkonen, E.; Brazma, A. A global map of human gene expression. Nat. Biotechnol. 2010, 28, 322–324. [Google Scholar] [CrossRef]
- Prakash, N. Developmental pathways linked to the vulnerability of adult midbrain dopaminergic neurons to neurodegeneration. Front. Mol. Neurosci. 2022, 15, 1071731. [Google Scholar] [CrossRef]
- Balestrini, S.; Milh, M.; Castiglioni, C.; Lüthy, K.; Finelli, M.J.; Verstreken, P.; Cardon, A.; Stražišar, B.G.; Holder, J.L., Jr.; Lesca, G.; et al. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 2016, 87, 77–85. [Google Scholar] [CrossRef]
- Zimmern, V.; Riant, F.; Roze, E.; Ranza, E.; Lehmann-Horn, F.; de Bellescize, J.; Ville, D.; Lesca, G.; Korff, C.M. Infantile-Onset Paroxysmal Movement Disorder and Episodic Ataxia Associated with a TBC1D24 Mutation. Neuropediatrics 2019, 50, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Steel, D.; Heim, J.; Kruer, M.C.; Sanchis Juan, A.; Raymond, L.F.; Eunson, P.; Kurian, M.A. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov. Disord. 2020, 35, 372–373. [Google Scholar] [CrossRef]
- Banuelos, E.; Ramsey, K.; Belnap, N.; Krishnan, M.; Balak, C.; Szelinger, S.; Siniard, A.L.; Russell, M.; Richholt, R.; De Both, M.; et al. Case Report: Novel mutations in TBC1D24 are associated with autosomal dominant tonic- clonic and myoclonic epilepsy and recessive Parkinsonism, psychosis, and intellectual disability. F1000Res 2017, 6, 553. [Google Scholar] [CrossRef] [PubMed]
- Falace, A.; Filipello, F.; La Padula, V.; Vanni, N.; Madia, F.; De Pietri Tonelli, D.; de Falco, F.A.; Striano, P.; Dagna Bricarelli, F.; Minetti, C.; et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 2010, 87, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Milh, M.; Falace, A.; Villeneuve, N.; Vanni, N.; Cacciagli, P.; Assereto, S.; Nabbout, R.; Benfenati, F.; Zara, F.; Chabrol, B.; et al. Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum. Mutat. 2013, 34, 869–872. [Google Scholar] [CrossRef]
- Murphy, K.C.; Volkert, M.R. Structural/functional analysis of the human OXR1 protein: Identification of exon 8 as the anti-oxidant encoding function. BMC Mol. Biol. 2012, 13, 26. [Google Scholar] [CrossRef]
- Madeo, M.; Stewart, M.; Sun, Y.; Sahir, N.; Wiethoff, S.; Chandrasekar, I.; Yarrow, A.; Rosenfeld, J.A.; Yang, Y.; Cordeiro, D.; et al. Loss-of-Function Mutations in FRRS1L Lead to an Epileptic- Dyskinetic Encephalopathy. Am. J. Hum. Genet. 2016, 98, 1249–1255. [Google Scholar] [CrossRef]
- Saadeldin, I.Y.; Al-Tala, S.M. Coexistence of epileptic encephalopathy with continuous spike-and-wave during sleep, atypical benign partial epilepsy, and fixation-off sensitivity in two siblings. Epilepsy Behav. 2011, 20, 116–122. [Google Scholar] [CrossRef]
- Hadi, D.A.; Mohamed, A.R.; Rethanavelu, K.; Khoo, T.B. Clonic seizures, continuousspikes-and-waves during slow sleep, choreoathetosis and response to sulthiame in a child with FRRS1L encephalopathy. Brain. Dev. 2022, 44, 44–49. [Google Scholar] [CrossRef]
- Shaheen, R.; Al Tala, S.; Ewida, N.; Abouelhoda, M.; Alkuraya, F.S. Epileptic encephalopathy with continuous spike-and-wave during sleep maps to a homozygous truncating mutation in AMPA receptor component FRRS1L. Clin. Genet. 2016, 90, 282–283. [Google Scholar] [CrossRef]
- Stewart, M.; Lau, P.; Banks, G.; Bains, R.S.; Castroflorio, E.; Oliver, P.L.; Dixon, C.L.; Kruer, M.C.; Kullmann, D.M.; Acevedo-Arozena, A.; et al. Loss of Frrs1l disrupts synaptic AMPA receptor function, and results in neurodevelopmental, motor, cognitive and electrographical abnormalities. Dis. Model. Mech. 2019, 12, dmm036806. [Google Scholar] [CrossRef]
- Brechet, A.; Buchert, R.; Schwenk, J.; Boudkkazi, S.; Zolles, G.; Siquier-Pernet, K.; Schaber, I.; Bildl, W.; Saadi, A.; Bole-Feysot, C.; et al. AMPA-receptor specific biogenesis complexes control synaptic transmission and intellectual ability. Nat. Commun. 2017, 8, 15910. [Google Scholar] [CrossRef]
- McNamara, J.O. Emerging insights into the genesis of epilepsy. Nature 1999, 399, A15–A22. [Google Scholar] [CrossRef] [PubMed]
- Gautam, V.; Rawat, K.; Sandhu, A.; Kumari, P.; Singh, N.; Saha, L. An insight into crosstalk among multiple signaling pathways contributing to epileptogenesis. Eur. J. Pharmacol. 2021, 910, 174469. [Google Scholar] [CrossRef]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef]
- Egbenya, D.L.; Aidoo, E.; Kyei, G. Glutamate receptors in brain development. Childs Nerv. Syst. 2021, 37, 2753–2758. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Butcher, N.J.; Kiehl, T.R.; Hazrati, L.N.; Chow, E.W.; Rogaeva, E.; Lang, A.E.; Bassett, A.S. Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: Identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol. 2013, 70, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Zaleski, C.; Bassett, A.S.; Tam, K.; Shugar, A.L.; Chow, E.W.; McPherson, E. The co- occurrence of early onset Parkinson disease and 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2009, 149A, 525–528. [Google Scholar] [CrossRef]
- Booij, J.; van Amelsvoort, T.; Boot, E. Co-occurrence of early-onset Parkinson disease and 22q11.2 deletion syndrome: Potential role for dopamine transporter imaging. Am. J. Med. Genet. A 2010, 152A, 2937–2938. [Google Scholar] [CrossRef]
- Rehman, A.F.; Dhamija, R.; Williams, E.S.; Barrett, M.J. 22q11.2 deletion syndrome presenting with early-onset Parkinson’s disease. Mov. Disord. 2015, 30, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Oki, M.; Hori, S.; Asayama, S.; Wate, R.; Kaneko, S.; Kusaka, H. Early-onset Parkinson’s Disease Associated with Chromosome 22q11.2 Deletion Syndrome. Intern. Med. 2016, 55, 303–305. [Google Scholar] [CrossRef]
- Pollard, R.; Hannan, M.; Tanabe, J.; Berman, B.D. Early-onset Parkinson disease leading to diagnosis of 22q11.2 deletion syndrome. Park. Relat. Disord. 2016, 25, 110–111. [Google Scholar] [CrossRef]
- Mok, K.Y.; Sheerin, U.; Simón-Sánchez, J.; Salaka, A.; Chester, L.; Escott-Price, V.; Mantripragada, K.; Doherty, K.M.; Noyce, A.J.; Mencacci, N.E.; et al. Deletions at 22q11.2 in idiopathic Parkinson’s disease: A combined analysis of genome-wide association data. Lancet Neurol. 2016, 15, 585–596. [Google Scholar] [CrossRef]
- Fanella, M.; Frascarelli, M.; Lambiase, C.; Morano, A.; Unolt, M.; Liberati, N.; Fattouch, J.; Buzzanca, A.; Accinni, T.; Ceccanti, M.; et al. Myoclonic epilepsy, Parkinsonism, schizophrenia and left-handedness as common neuropsychiatric features in 22q11.2 deletion syndrome. J. Med. Genet. 2020, 57, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Boot, E.; Butcher, N.J.; Udow, S.; Marras, C.; Mok, K.Y.; Kaneko, S.; Barrett, M.J.; Prontera, P.; Berman, B.D.; Masellis, M.; et al. Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 2018, 90, e2059–e2067. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk—Database: Prediction of possible miRNA binding sites by “waling” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–884. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Maynard, T.M.; Meechan, D.W.; Dudevoir, M.L.; Gopalakrishna, D.; Peters, A.Z.; Heindel, C.C.; Sugimoto, T.J.; Wu, Y.; Lieberman, J.A.; Lamantia, A.S. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell Neurosci. 2008, 39, 439–451. [Google Scholar] [CrossRef]
- Maynard, T.M.; Haskell, G.T.; Peters, A.Z.; Sikich, L.; Lieberman, J.A.; LaMantia, A.S. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 14433–14438. [Google Scholar] [CrossRef] [PubMed]
- Brzustowicz, L.M.; Bassett, A.S. miRNA-mediated risk for schizophrenia in 22q11.2 deletion syndrome. Front. Genet. 2012, 3, 291. [Google Scholar] [CrossRef] [PubMed]
- Butcher, N.J.; Merico, D.; Zarrei, M.; Ogura, L.; Marshall, C.R.; Chow, E.W.C.; Lang, A.E.; Scherer, S.W.; Bassett, A.S. Whole-genome sequencing suggests mechanisms for 22q11.2 deletion-associated Parkinson’s disease. PLoS ONE 2017, 12, e0173944. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chai, H.; DiAdamo, A.; Grommisch, B.; Wen, J.; Zhang, H.; Li, P. Genotype-Phenotype Correlations for Putative Haploinsufficient Genes in Deletions of 6q26-q27: Report of Eight Patients and Review of Literature. Glob. Med. Genet. 2022, 9, 166–174. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2023, 84, 101817. [Google Scholar] [CrossRef]
- Vieregge, P.; Ziemens, G.; Piosinski, A.; Freudenberg, M.; Kömpf, D. Parkinsonian features in advanced Down’s syndrome. J. Neural Transm. Suppl. 1991, 33, 119–124. [Google Scholar]
- Bach, J.P.; Sommer, N.; Moller, J.C.; Oertel, W.H.; Dodel, R.; Gasser, T. Parkinson’s syndrome in a young patient with Klinefelter’s syndrome: A case report. Mov. Disord. 2008, 23, 771–772. [Google Scholar] [CrossRef]
- Grosso, S.; Farnetani, M.A.; Di Bartolo, R.M.; Berardi, R.; Pucci, L.; Mostardini, R.; Anichini, C.; Bartalini, G.; Galimberti, D.; Morgese, G.; et al. Electroencephalographic and epileptic patterns in X chromosome anomalies. J. Clin. Neurophysiol. 2004, 21, 249–253. [Google Scholar] [CrossRef]
- Gavaret, M.; Bartolomei, F.; Daquin, G.; Gastaut, J.-L. Absence-Epilepsy and Klinefelter Syndrome. J. Epilepsy 1997, 10, 12–14. [Google Scholar] [CrossRef]
- Namihira, T.; Hattori, N.; Shiroma, S.; Miyazato, Y. Autosomal recessive juvenile Parkinson’s disease with partial trisomy of chromosome 6q syndrome: A case report. Psychiatry Clin. Neurosci. 2004, 58, 672–673. [Google Scholar] [CrossRef]
- Garraux, G.; Caberg, J.H.; Vanbellinghen, J.F.; Jamar, M.; Bours, V.; Moonen, G.; Dive, D. Partial trisomy 4q associated with young-onset dopa-responsive Parkinsonism. Arch. Neurol. 2012, 69, 398–400. [Google Scholar] [CrossRef]
- Carvalho, V.; Ferreira, J.J.; Correia Guedes, L. Tremor and Parkinsonism in Chromosomopathies—A Systematic Review. Mov. Disord. 2021, 36, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [PubMed]
- Peron, A.; Novara, F.; La Briola, F.; Merati, E.; Giannusa, E.; Segalini, E.; Anniballi, G.; Vignoli, A.; Ciccone, R.; Canevini, M.P. Missense variants in the Arg206 residue of HNRNPH2: Further evidence of causality and expansion of the phenotype. Am. J. Med. Genet. A 2020, 182, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Liu, A.; Mandelstam, S.; Schneider, A.; Lacroix, A.; Zemel, M.; McMahon, J.M.; Bello-Espinosa, L.; Mackay, M.; Wallace, G.; et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia 2018, 59, e5–e13. [Google Scholar] [CrossRef] [PubMed]
- Paudel, R.; Li, A.; Wiethoff, S.; Bandopadhyay, R.; Bhatia, K.; de Silva, R.; Houlden, H.; Holton, J.L. Neuropathology of Beta-propeller protein associated neurodegeneration (BPAN): A new tauopathy. Acta. Neuropathol. Commun. 2015, 3, 39. [Google Scholar] [CrossRef]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–450. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Q.; Chen, X.; Zeng, Q.; Shao, Y.; Fang, H.; Liao, X.; Li, H.S.; Liu, M.G.; Xu, T.L.; et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 2020, 16, 531–547. [Google Scholar] [CrossRef]
- Mollereau, B.; Walter, L. Is WDR45 the missing link for ER stress -induced autophagy in beta-propeller associated neurodegeneration? Autophagy 2019, 15, 2163–2164. [Google Scholar] [CrossRef]
- Gonzalez-Alegre, P.; Afifi, A.K. Clinical characteristics of childhood-onset (juvenile) Huntington disease: Report of 12 patients and review of the literature. J. Child Neurol. 2006, 21, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Richards, F.H.; Jones, K.J.; Ryan, M.M. Juvenile Huntington disease. J. Paediatr. Child Health 2006, 42, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Siesling, S.; Vegler-van der Vlis, M.; Roos, R.A. Juvenile Huntington disease in the Netherlands. Pediatr. Neurol. 1997, 17, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A. Genetic testing of children at risk for Huntington’s disease. US Huntington Disease Genetic Testing Group. Neurology 1997, 49, 1048–1053. [Google Scholar] [CrossRef]
- Weiner, W.J.; Lang, A.E. Movement Disorders: A Comprehensive Survey; Futura Publishing: Mount Kisco, NY, USA, 1989; pp. 293–346. [Google Scholar]
- Menkes, J.H. Huntington’s disease: Finding the gene and after. Pediatr. Neurol. 1988, 4, 73–78. [Google Scholar] [CrossRef]
- Dewhurst, K.; Oliver, J.E.; McKnight, A.L. Socio-psychiatric consequences of Huntington’s disease. Br. J. Psychiatry 1970, 116, 255–258. [Google Scholar] [CrossRef]
- Osborne, J.P.; Munson, P.; Burman, D. Huntington’s chorea. Report of 3 cases and review of literature. Arch. Dis. Child 1982, 57, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Seneca, S.; Fagnart, D.; Keymolen, K.; Lissens, W.; Hasaerts, D.; Debulpaep, S.; Desprechins, B.; Liebaers, I.; De Meirleir, L. Early onset Huntington disease: A neuronal degeneration syndrome. Eur. J. Pediatr. 2004, 163, 717–721. [Google Scholar] [CrossRef]
- Lee, Y.C.; Durr, A.; Majczenko, K.; Huang, Y.H.; Liu, Y.C.; Lien, C.C.; Tsai, P.C.; Ichikawa, Y.; Goto, J.; Monin, M.L.; et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Duarri, A.; Jezierska, J.; Fokkens, M.; Meijer, M.; Schelhaas, H.J.; den Dunnen, W.F.; van Dijk, F.; Verschuuren-Bemelmans, C.; Hageman, G.; van de Vlies, P.; et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 2012, 72, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Duarri, A.; Nibbeling, E.; Fokkens, M.R.; Meijer, M.; Boddeke, E.; Lagrange, E.; Stevanin, G.; Brice, A.; Durr, A.; Verbeek, D.S. The L450F [Corrected] mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together. Neurogenetics 2013, 14, 257–258. [Google Scholar] [CrossRef]
- Pollini, L.; Galosi, S.; Tolve, M.; Caputi, C.; Carducci, C.; Angeloni, A.; Leuzzi, V. KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. Int. J. Mol. Sci. 2020, 21, 5802. [Google Scholar] [CrossRef]
- Poorkaj, P.; Raskind, W.H.; Leverenz, J.B.; Matsushita, M.; Zabetian, C.P.; Samii, A.; Kim, S.; Gazi, N.; Nutt, J.G.; Wolff, J.; et al. A novel X-linked four-repeat tauopathy with Parkinsonism and spasticity. Mov. Disord. 2010, 25, 1409–1417. [Google Scholar] [CrossRef]
- Hedera, P.; Alvarado, D.; Beydoun, A.; Fink, J.K. Novel mental retardation-epilepsy syndrome linked to Xp21.1-p11.4. Ann. Neurol. 2002, 51, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.V.; Vengoechea, J.; Sahaya, K.; Virmani, T. A splice site mutation in ATP6AP2 causes X-linked intellectual disability, epilepsy, and Parkinsonism. Park. Relat. Disord. 2015, 21, 1473–1475. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin uncoating co-chaperone auxilin, is associated with juvenile Parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed]
- Köroğlu, Ç.; Baysal, L.; Cetinkaya, M.; Karasoy, H.; Tolun, A. DNAJC6 is responsible for juvenile Parkinsonism with phenotypic variability. Park. Relat. Disord. 2013, 19, 320–324. [Google Scholar] [CrossRef]
- Vauthier, V.; Jaillard, S.; Journel, H.; Dubourg, C.; Jockers, R.; Dam, J. Homozygous deletion of an 80 kb region comprising part of DNAJC6 and LEPR genes on chromosome 1P31.3 is associated with early onset obesity, mental retardation and epilepsy. Mol. Genet. Metab. 2012, 106, 345–350. [Google Scholar] [CrossRef]
- Paesmans, J.; Martin, E.; Deckers, B.; Berghmans, M.; Sethi, R.; Loeys, Y.; Pardon, E.; Steyaert, J.; Verstreken, P.; Galicia, C.; et al. A structure of substrate-bound Synaptojanin1 provides new insights in its mechanism and the effect of disease mutations. Elife 2020, 9, e64922. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; Cai, Y.; Jardel, C.; Jansen, A.C.; Cao, M.; May, P.; Djémié, T.; Hachon Le Camus, C.; Keymolen, K.; Deconinck, T.; et al. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain 2016, 139, 2420–2430. [Google Scholar] [CrossRef]
- Dyment, D.A.; Smith, A.C.; Humphreys, P.; Schwartzentruber, J.; Beaulieu, C.L.; FORGE Canada Consortium; Bulman, D.E.; Majewski, J.; Woulfe, J.; Michaud, J.; et al. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol. Aging 2015, 36, e1222.e1–e1222.e5. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Tétreault, M.; Beaulieu, C.L.; Hartley, T.; Ferreira, P.; Chardon, J.W.; Marcadier, J.; Sawyer, S.L.; Mosca, S.J.; Innes, A.M.; et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: A retrospective study. Clin. Genet. 2015, 88, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; Di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The Sac1domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef]
- Olgiati, S.; De Rosa, A.; Quadri, M.; Criscuolo, C.; Breedveld, G.J.; Picillo, M.; Pappatà, S.; Quarantelli, M.; Barone, P.; De Michele, G.; et al. PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 2014, 15, 183–188. [Google Scholar] [CrossRef]
- Ben Romdhan, S.; Sakka, S.; Farhat, N.; Triki, S.; Dammak, M.; Mhiri, C. A Novel SYNJ1 Mutation in a Tunisian Family with Juvenile Parkinson’s Disease Associated with Epilepsy. J. Mol. Neurosci. 2018, 66, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Chen, S.; Le, W.D. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, A.; Zech, M.; Sigafoos, A.N.; Clark, K.J.; Dincer, Y.; Wagner, M.; Humberson, J.B.; Green, S.; van Gassen, K.; et al. De novo variants of NR4A2 are associated with neurodevelopmental disorder and epilepsy. Genet. Med. 2020, 22, 1413–1417. [Google Scholar] [CrossRef]
- Wirth, T.; Mariani, L.L.; Bergant, G.; Baulac, M.; Habert, M.O.; Drouot, N.; Ollivier, E.; Hodžić, A.; Rudolf, G.; Nitschke, P.; et al. Loss-of-Function Mutations in NR4A2 Cause Dopa-Responsive Dystonia Parkinsonism. Mov. Disord. 2020, 35, 880–885. [Google Scholar] [CrossRef]
- Winter, B.; Krämer, J.; Meinhardt, T.; Berner, D.; Alt, K.; Wenzel, M.; Winkelmann, J.; Zech, M. NR4A2 and Dystonia with Dopa Responsiveness. Mov. Disord. 2021, 36, 2203–2204. [Google Scholar] [CrossRef]
- Ramos, L.L.P.; Monteiro, F.P.; Sampaio, L.P.B.; Costa, L.A.; Ribeiro, M.D.O.; Freitas, E.L.; Kitajima, J.P.; Kok, F. Heterozygous loss of function of NR4A2 is associated with intellectual deficiency, rolandic epilepsy, and language impairment. Clin. Case Rep. 2019, 7, 1582–1584. [Google Scholar] [CrossRef]
- Mignogna, M.L.; Musardo, S.; Ranieri, G.; Gelmini, S.; Espinosa, P.; Marra, P.; Belloli, S.; Murtaj, V.; Moresco, R.M.; Bellone, C.; et al. RAB39B-mediated trafficking of the GluA2-AMPAR subunit controls dendritic spine maturation and intellectual disability-related behaviour. Mol. Psychiatry 2021, 26, 6531–6549. [Google Scholar] [CrossRef]
- Mignogna, M.L.; Giannandrea, M.; Gurgone, A.; Fanelli, F.; Raimondi, F.; Mapelli, L.; Bassani, S.; Fang, H.; Van Anken, E.; Alessio, M.; et al. The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat. Commun. 2015, 6, 6504. [Google Scholar] [CrossRef]
- Wilson, G.R.; Sim, J.C.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef]
- Giannandrea, M.; Bianchi, V.; Mignogna, M.L.; Sirri, A.; Carrabino, S.; D’Elia, E.; Vecellio, M.; Russo, S.; Cogliati, F.; Larizza, L.; et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet. 2010, 86, 185–195. [Google Scholar] [CrossRef]
- Russo, S.; Cogliati, F.; Cavalleri, F.; Cassitto, M.G.; Giglioli, R.; Toniolo, D.; Casari, G.; Larizza, L. Mapping to distal Xq28 of nonspecific X-linked mental retardation MRX72: Linkage analysis and clinical findings in a three-generation Sardinian family. Am. J. Med. Genet. 2000, 94, 376–382. [Google Scholar] [CrossRef]
- Laxova, R.; Brown, E.S.; Hogan, K.; Hecox, K.; Opitz, J.M. An X-linked recessive basal ganglia disorder with mental retardation. Am. J. Med. Genet. 1985, 21, 681–689. [Google Scholar] [CrossRef]
- Kaur, S.; Christodoulou, J. MECP2 Disorders. 2001 Oct 3 [updated 2019 Sep 19]; In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Inuzuka, L.M.; Guerra-Peixe, M.; Macedo-Souza, L.I.; Pedreira, C.C.; Gurgel-Giannetti, J.; Monteiro, F.P.; Ramos, L.; Costa, L.A.; Crippa, A.C.S.; Lourenco, C.M.; et al. MECP2-related conditions in males: A systematic literature review and 8 additional cases. Eur. J. Paediatr. Neurol. 2021, 34, 7–13. [Google Scholar] [CrossRef]
- John, A.; Ng-Cordell, E.; Hanna, N.; Brkic, D.; Baker, K. The neurodevelopmental spectrum of synaptic vesicle cycling disorders. J. Neurochem. 2021, 157, 208–228. [Google Scholar] [CrossRef]
- Abramov, D.; Guiberson, N.G.L.; Burré, J. STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies. J. Neurochem. 2021, 157, 165–178. [Google Scholar] [CrossRef]
- Melland, H.; Carr, E.M.; Gordon, S.L. Disorders of synaptic vesicle fusion machinery. J. Neurochem. 2021, 157, 130–164. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Bougea, A. Synuclein in neurodegeneration. Adv. Clin. Chem. 2021, 103, 97–134. [Google Scholar] [PubMed]
- Runwal, G.; Edwards, R.H. The membrane interactions of synuclein: Physiology and pathology. Annu. Rev. Pathol. 2021, 16, 465–485. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Nassif, M.; Hetz, C. Autophagy impairment: A crossroad between neurodegeneration and tauopathies. BMC Biol. 2012, 10, 78. [Google Scholar] [CrossRef]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.E.; Ruiz-Lopez, M.; Dalmau, J.; Erro, R.; Privitera, M.; Andrade, D.; Fasano, A. Seizures and movement disorders: Phenomenology, diagnostic challenges and therapeutic approaches. J. Neurol. Neurosurg. Psychiatry 2019, 90, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Condition Name | Epilepsy | Parkinsonism | Additional Developmental and Psychiatric Features |
|---|---|---|---|
| 22q11.2 deletion syndrome | Increased risk of seizures (provoked, unprovoked, focal or generalized) | Teenage to adult years | Psychiatric symptoms, ADHD, ASD, ID |
| 6q26–q27 Deletion | Increased risk of (drug-responsive) seizures | Single report | ID, ASD, ADHD |
| Trisomy 21 | Increased risk of epilepsy, West syndrome | EOP | ID, ASD, ADHD, increased risk of dementia |
| Klinefelter syndrome | Increased risk of epilepsy | Single report (27 y) | ID |
| Partial 6q syndrome | Not known to be associated with increased risk of epilepsy | Single report (35 y) | ID |
| Partial 4q syndrome | Not known to be associated with increased risk of epilepsy | Single report (30 y) | ID |
| Inheritance | Epilepsy | Parkinsonism | Additional Features | |
|---|---|---|---|---|
| WDR45 | XL | Childhood | Early adulthood | DD, regression, cognitive decline |
| IT15 | CAG repeats expansion | Childhood | Childhood | Aggressive, hyperactive, oppositional behavior |
| Inheritance | Epilepsy, Age at Onset | Parkinsonism, Age at Onset | Developmental and Additional Neurological Features | |
|---|---|---|---|---|
| KCND3 | AD | Infancy-childhood (early-onset phenotype—see text) | Second to fifth decade | Ataxia, oculomotor disorders, possible ID, NDD, myoclonus, dystonia, tremor, peripheral neuropathy, pyramidal signs |
| ATP6AP2 | XL | Infancy | First to sixth decade | Mild-to-moderate ID, ataxia, scholiosis |
| DNAJC6 | AR | 1–5 y | First to third decade | Early-onset tremor, shuffling gait, and/or bradykinesia |
| SYNJ1 | Infancy to adolescence | Second to third decade | Mild cognitive impairment | |
| NR4A2 | AD | Childhood to early adulthood | Infantile-onset to early-onset parkinsonism | Poor feeding, gastrointestinal symptoms, normal motor but delayed speech development |
| RAB39B | XL | Early childhood | Second to fifth decade | ID, ASD, macrocephaly, tremors, choreoathethoid movements and upper limbs rigidity (10 y) |
| MECP2 pathogenic variants in males | XL | Possible (with childhood onset) in XLMR13 phenotype; absent in PPM-X phenotype | After the first decade (PPM-X phenotype) | Mild-to-severe DD or ID, mood and behavioral problems, psychosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnoli, C.; Fusco, C.; Pisani, F. Pediatric-Onset Epilepsy and Developmental Epileptic Encephalopathies Followed by Early-Onset Parkinsonism. Int. J. Mol. Sci. 2023, 24, 3796. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043796
Spagnoli C, Fusco C, Pisani F. Pediatric-Onset Epilepsy and Developmental Epileptic Encephalopathies Followed by Early-Onset Parkinsonism. International Journal of Molecular Sciences. 2023; 24(4):3796. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043796
Chicago/Turabian StyleSpagnoli, Carlotta, Carlo Fusco, and Francesco Pisani. 2023. "Pediatric-Onset Epilepsy and Developmental Epileptic Encephalopathies Followed by Early-Onset Parkinsonism" International Journal of Molecular Sciences 24, no. 4: 3796. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043796