
A Competition between Relative Stability and Binding Energy in Caffeine Phenyl-Glucose Aggregates: Implications in Biological Mechanisms
 , , , , and
, , , , and
Abstract
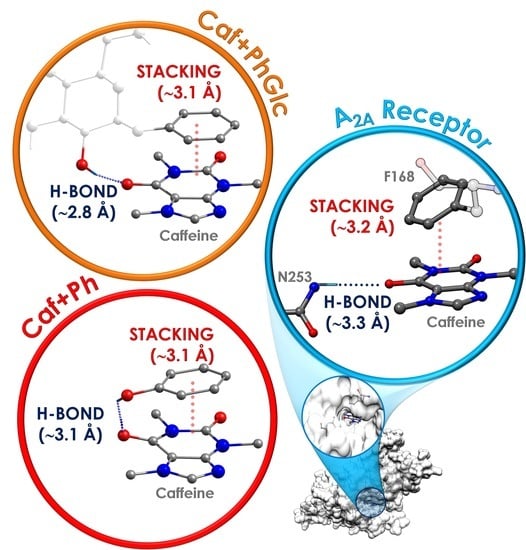
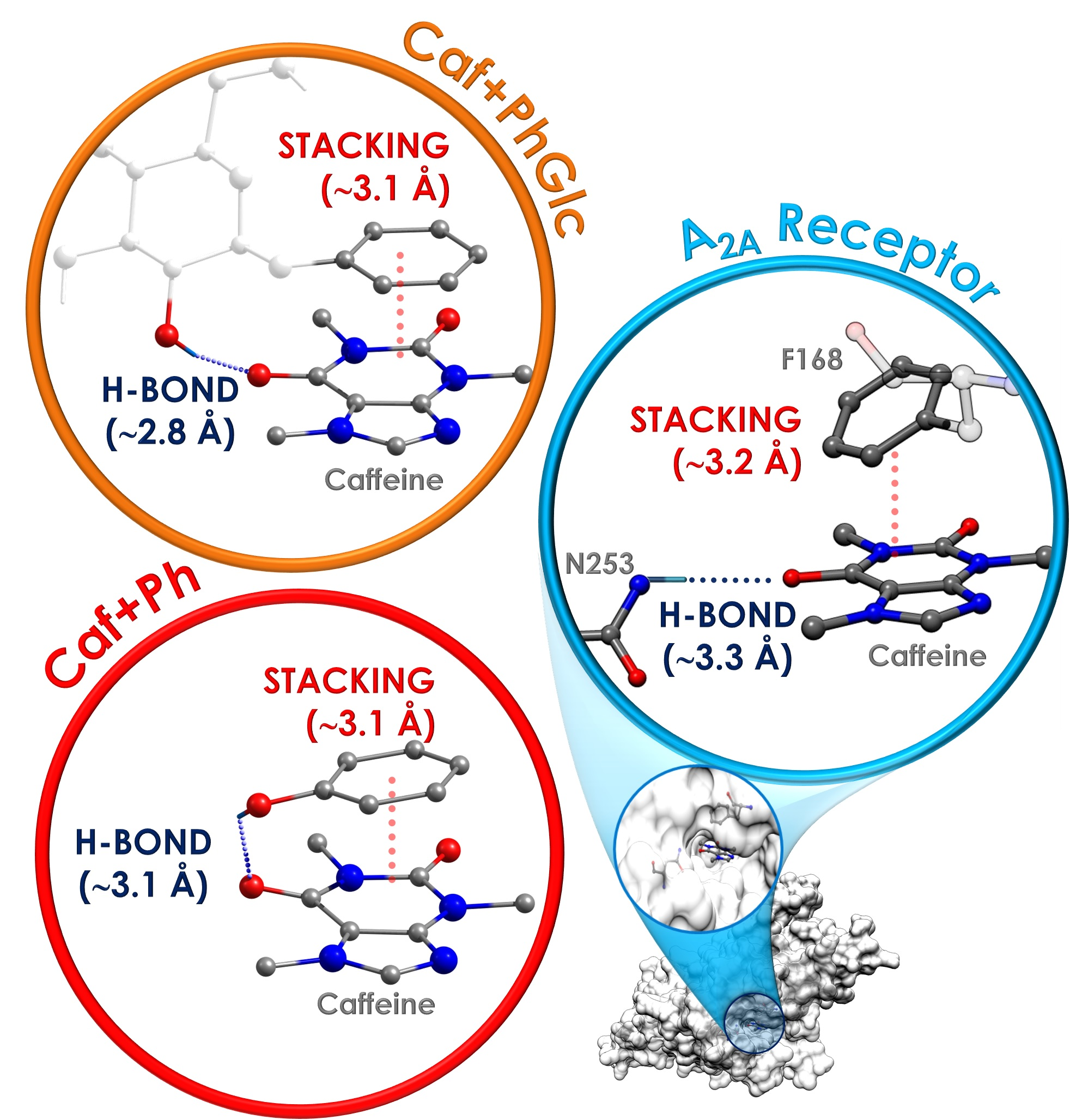
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
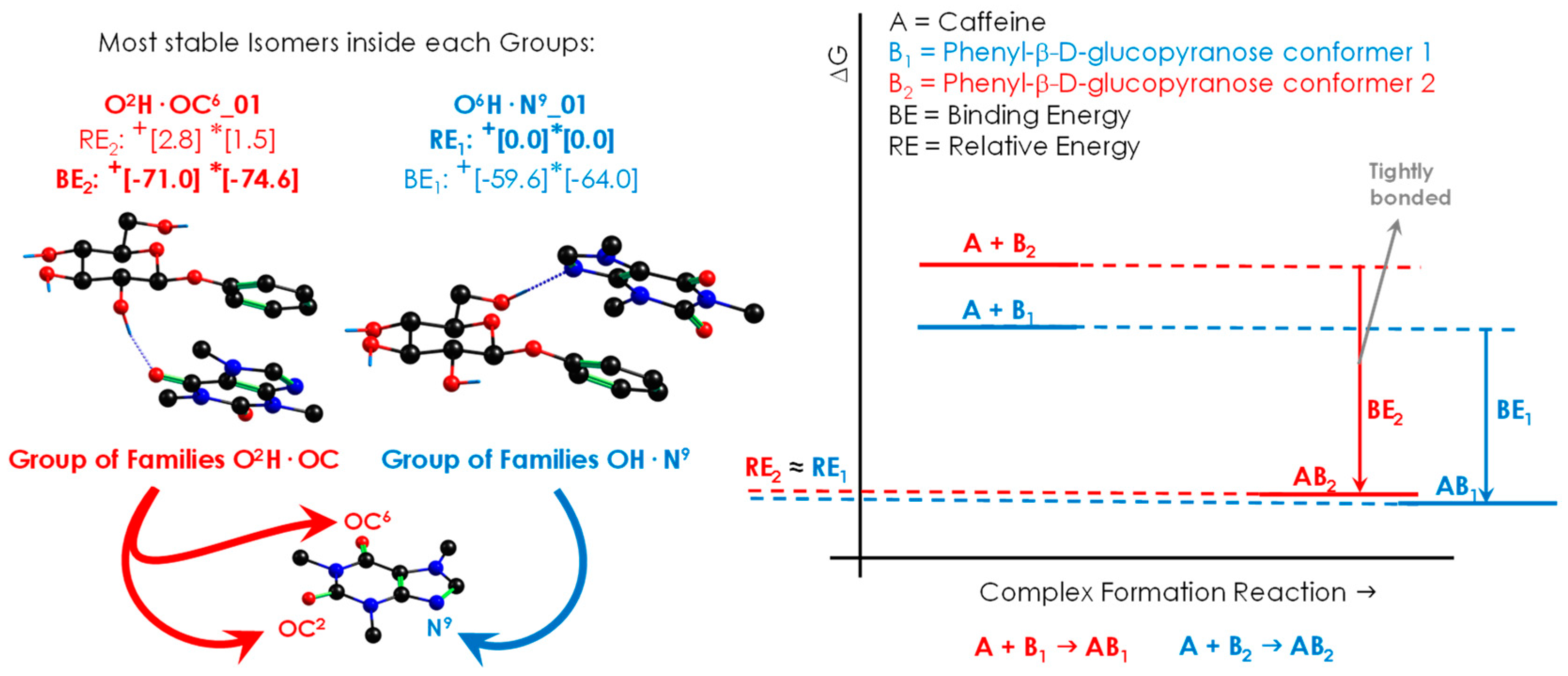
2. Results
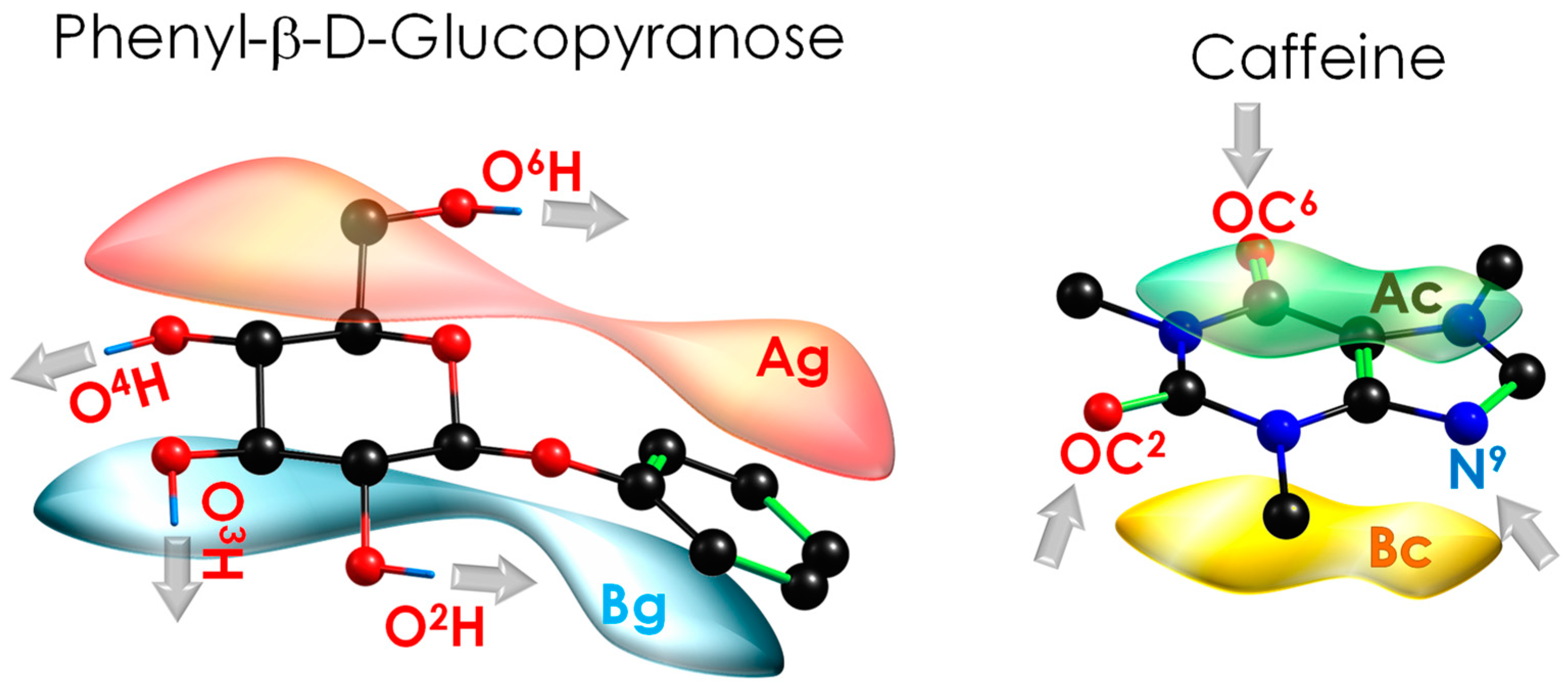
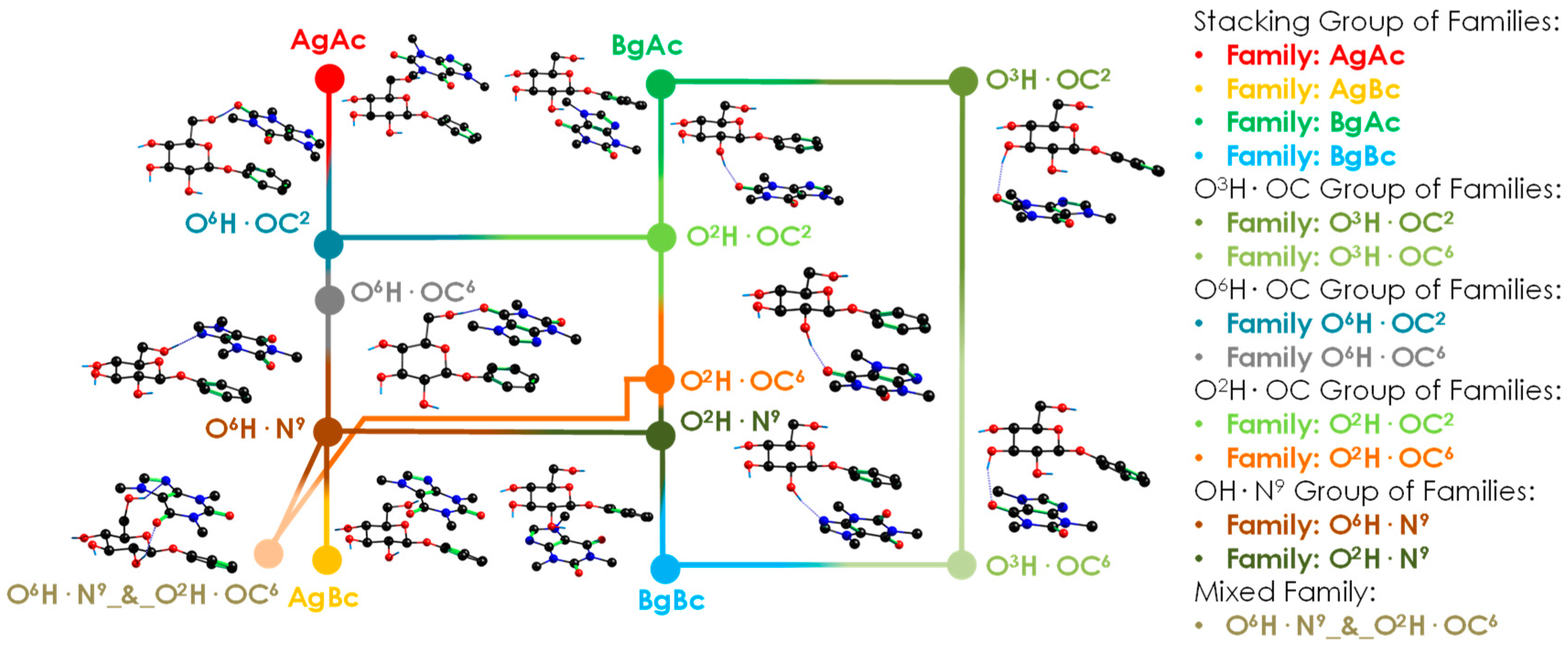
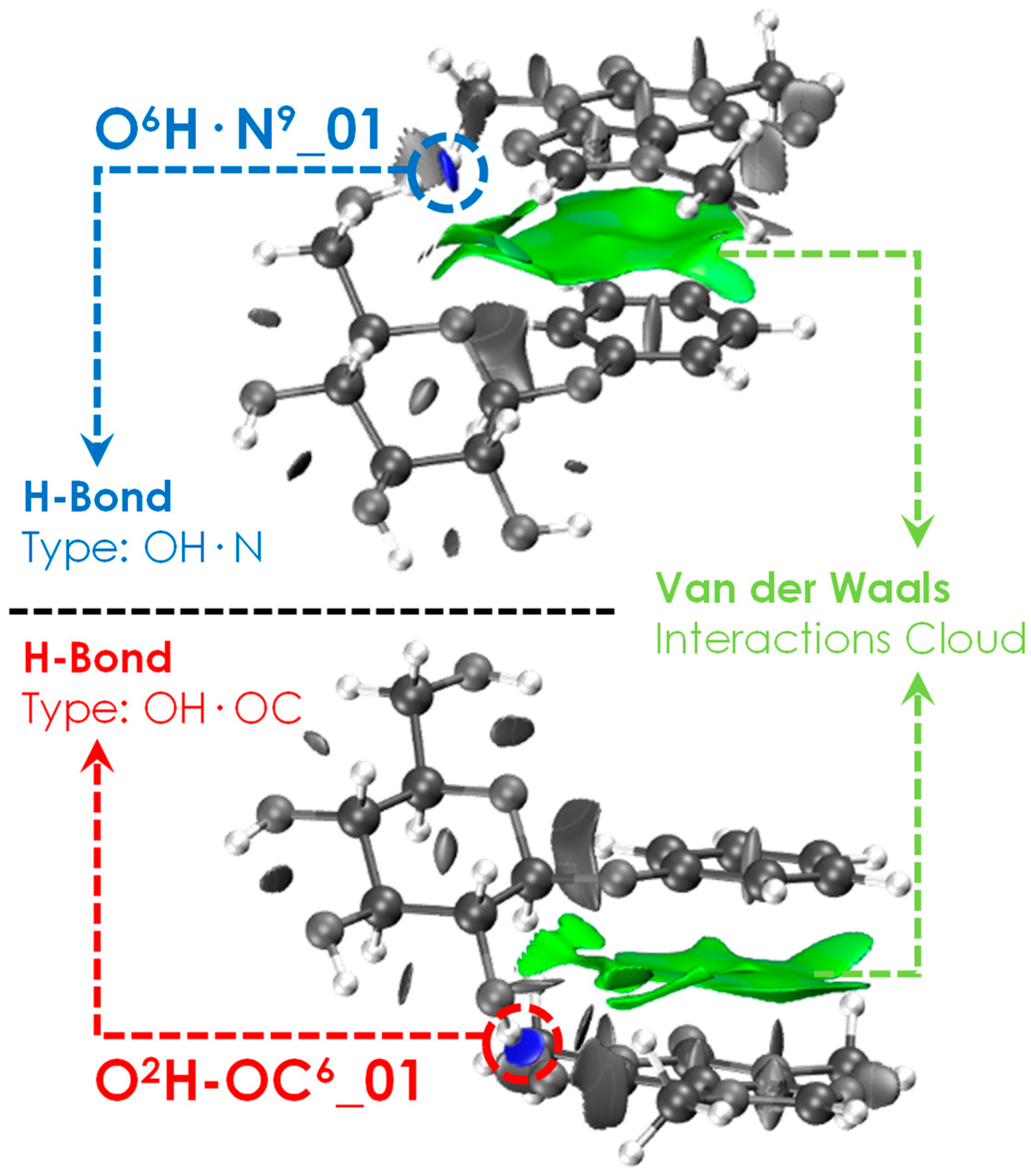
2.1. Exploration of the Intermolecular Potential Energy Surface (iPES)
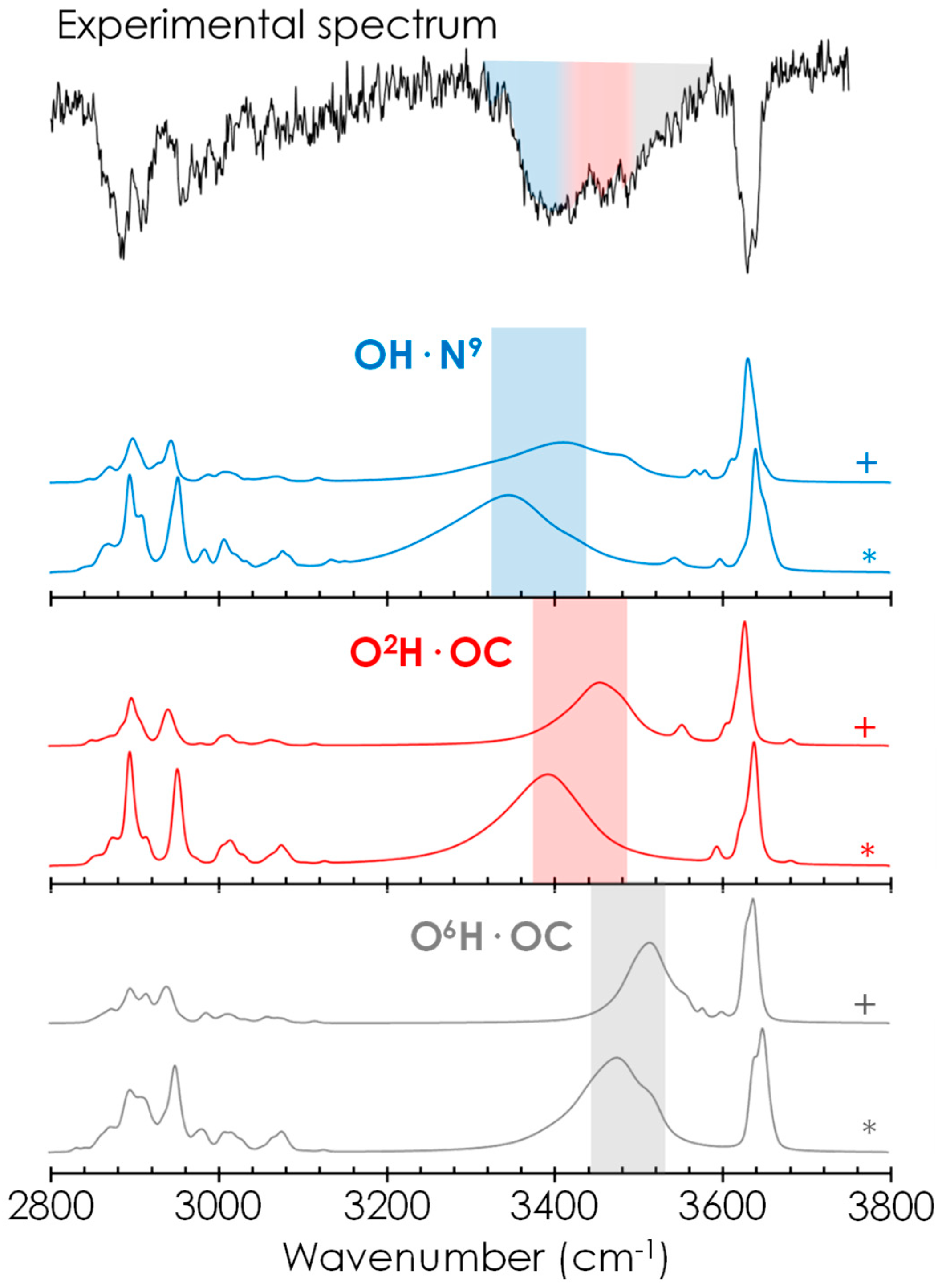
2.2. Spectroscopic Characterisation of Caf+PhGlc Complex
3. Discussion
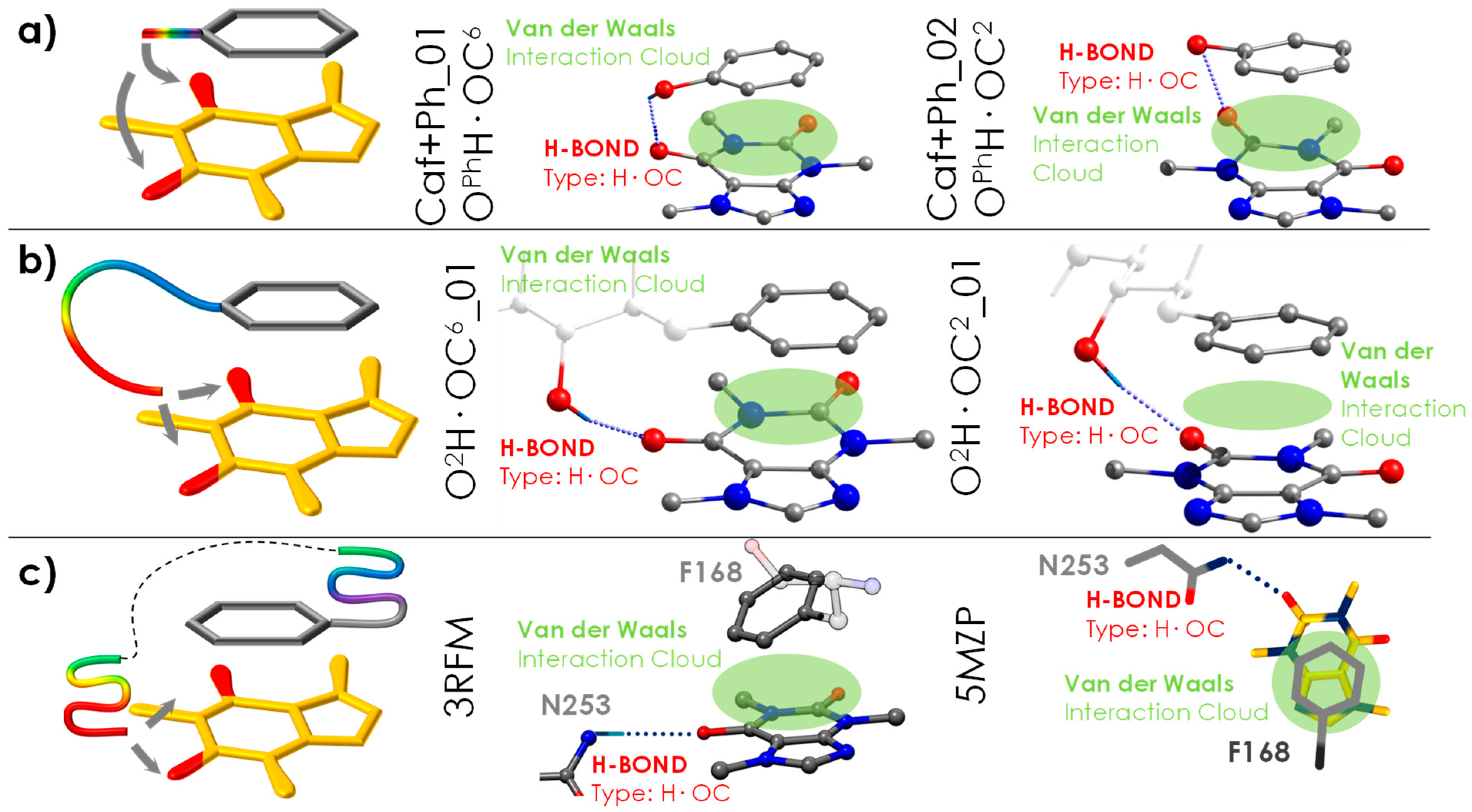
3.1. General Tendency in the Interaction with Caffeine
3.2. General Tendency and the Similarities with the Interaction inside the A2A Receptor
4. Materials and Methods
4.1. Computations
4.2. Experimental Setup
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Astorino, T.A.; Alkadhi, K.A.; Addicott, M.; Farah, A.; Balssa, F.; Keast, R.S.J.; Chapman, R.F.; Acquas, E.; Reis, J.P.; Sen, D.J.; et al. Caffeine: Chemistry, Analysis, Function and Effects; Preedy, V.R., Ed.; Food and Nutritional Components in Focus; The Royal Society of Chemistry: London, UK, 2012; ISBN 978-1-84973-367-0. [Google Scholar]
- Latosińska, J.N.; Latosińska, M.; Olejniczak, G.A.; Seliger, J.; Žagar, V. Topology of the Interactions Pattern in Pharmaceutically Relevant Polymorphs of Methylxanthines (Caffeine, Theobromine, and Theophiline): Combined Experimental (1H-14N Nuclear Quadrupole Double Resonance) and Computational (DFT and Hirshfeld-BASED) Study. J. Chem. Inf. Model. 2014, 54, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A.; Daval, J.L.; Debry, G. Caffeine and the Central Nervous System: Mechanisms of Action, Biochemical, Metabolic and Psychostimulant Effects. Brain Res. Rev. 1992, 17, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, S.H.; Katims, J.J.; Annau, Z.; Bruns, R.F.; Daly, J.W. Adenosine Receptors and Behavioral Actions of Methylxanthines. Proc. Natl. Acad. Sci. USA 1981, 78, 3260–3264. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.A.; Sebastiao, A.M. Caffeine and Adenosine. J. Alzheimer’s Dis. 2010, 20, S3–S15. [Google Scholar] [CrossRef] [Green Version]
- McLellan, T.M.; Caldwell, J.A.; Lieberman, H.R. A Review of Caffeine’s Effects on Cognitive, Physical and Occupational Performance. Neurosci. Biobehav. Rev. 2016, 71, 294–312. [Google Scholar] [CrossRef] [Green Version]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the Adenosine A 2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef]
- Pyrkov, T.V.; Pyrkova, D.V.; Balitskaya, E.D.; Efremov, R.G. The Role of Stacking Interactions in Complexes of Proteins with Adenine and Guanine Fragments of Ligands. Acta Nat. 2009, 1, 124–127. [Google Scholar] [CrossRef]
- Kataev, E.A.; Shumilova, T.A.; Fiedler, B.; Anacker, T.; Friedrich, J. Understanding Stacking Interactions between an Aromatic Ring and Nucleobases in Aqueous Solution: Experimental and Theoretical Study. J. Org. Chem. 2016, 81, 6505–6514. [Google Scholar] [CrossRef]
- Tsuzuki, S. CH/π Interactions. Annu. Rep. Prog. Chem.-Sect. C 2012, 108, 69–95. [Google Scholar] [CrossRef]
- Asensio, J.L.; Ardá, A.; Cañada, F.J.; Jiménez-Barbero, J. Carbohydrate-Aromatic Interactions. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavagnacco, L.; Engström, O.; Schnupf, U.; Saboungi, M.L.; Himmel, M.; Widmalm, G.; Cesàro, A.; Brady, J.W. Caffeine and Sugars Interact in Aqueous Solutions: A Simulation and NMR Study. J. Phys. Chem. B 2012, 116, 11701–11711. [Google Scholar] [CrossRef] [Green Version]
- Usabiaga, I.; Camiruaga, A.; Calabrese, C.; Maris, A.; Fernández, J.A. Exploring Caffeine–Phenol Interactions by the Inseparable Duet of Experimental and Theoretical Data. Chem.-A Eur. J. 2019, 25, 14230–14236. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.S.; Hobza, P. Gas-Phase Spectroscopy of Biomolecular Building Blocks. Annu. Rev. Phys. Chem. 2007, 58, 585–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijs, A.M.; Oomens, J. IR Spectroscopic Techniques to Study Isolated Biomolecules. In Gas-Phase IR Spectroscopy and Structure of Biological Molecules; Rijs, A.M., Oomens, J., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–42. ISBN 978-3-319-19204-8. [Google Scholar]
- Kim, D.; Kim, H.M.; Yang, K.Y.; Kim, S.K.; Kim, N.J. Molecular Beam Resonant Two-Photon Ionization Study of Caffeine and Its Hydrated Clusters. J. Chem. Phys. 2008, 128, 134310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Usabiaga, I.; González, J.; Arnáiz, P.F.; León, I.; Cocinero, E.J.; Fernández, J.A. Modeling the Tyrosine–Sugar Interactions in Supersonic Expansions: Glucopyranose–Phenol Clusters. Phys. Chem. Chem. Phys. 2016, 18, 12457–12465. [Google Scholar] [CrossRef] [Green Version]
- Usabiaga, I.; Camiruaga, A.; Insausti, A.; Çarçabal, P.; Cocinero, E.J.; León, I.; Fernández, J.A. Phenyl-β-D-Glucopyranoside and Phenyl-β-D-Galactopyranoside Dimers: Small Structural Differences but Very Different Interactions. Front. Phys. 2018, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Cocinero, E.J.; Stanca-Kaposta, E.C.; Davis, B.G.; Simons, J.P. Carbohydrate-Aromatic Interactions: A Computational and IR Spectroscopic Investigation of the Complex, Methyl α-l-Fucopyranoside · Toluene, Isolated in the Gas Phase. Chem. Phys. Lett. 2009, 471, 17–21. [Google Scholar] [CrossRef]
- Camiruaga, A.; Usabiaga, I.; Insausti, A.; León, I.; Fernández, J.A. Sugar-Peptidic Bond Interactions: Spectroscopic Characterization of a Model System. Phys. Chem. Chem. Phys. 2017, 19, 12013–12021. [Google Scholar] [CrossRef] [PubMed]
- Forsting, T.; Gottschalk, H.C.; Hartwig, B.; Mons, M.; Suhm, M.A. Correcting the Record: The Dimers and Trimers of Trans-N-Methylacetamide. Phys. Chem. Chem. Phys. 2017, 19, 10727–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, B.; Lange, M.; Poblotzki, A.; Medel, R.; Zehnacker, A.; Suhm, M.A. The Reduced Cohesion of Homoconfigurational 1,2-Diols. Phys. Chem. Chem. Phys. 2020, 22, 1122–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollipost, F.; Otto, K.E.; Suhm, M.A. A Symmetric Recognition Motif between Vicinal Diols: The Fourfold Grip in Ethylene Glycol Dimer. Angew. Chem. 2016, 128, 4667–4671. [Google Scholar] [CrossRef] [Green Version]
- Uriarte, I.; Insausti, A.; Cocinero, E.J.; Jabri, A.; Kleiner, I.; Mouhib, H.; Alkorta, I. Competing Dispersive Interactions: From Small Energy Differences to Large Structural Effects in Methyl Jasmonate and Zingerone. J. Phys. Chem. Lett. 2018, 9, 5906–5914. [Google Scholar] [CrossRef]
- Usabiaga, I.; Camiruaga, A.; Calabrese, C.; Veloso, A.; D’mello, V.C.; Wategaonkar, S.; Fernández, J.A. Exploration of the Theobromine-Water Dimer: Comparison with DNA Microhydration. Phys. Chem. Chem. Phys. 2020, 22, 15759–15768. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- MacroModel and SchrödingerRelease v4, 2019; Maestro Schrödinger Release 2023-1; Maestro, Schrödinger, LLC: New York, NY, USA, 2021.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V. Gaussian16 2016, Gaussian 16, Revision C.01. Available online: https://gaussian.com/citation/ (accessed on 24 January 2023).
- León, I.; Usabiaga, I.; Millán, J.; Cocinero, E.J.; Lesarri, A.; Fernández, J.A. Mimicking Anesthetic–Receptor Interactions in Jets: The Propofol–Isopropanol Cluster. Phys. Chem. Chem. Phys. 2014, 16, 16968–16975. [Google Scholar] [CrossRef]
- Camiruaga, A.; Usabiaga, I.; D’Mello, V.C.; García, G.A.; Wategaonkar, S.; Fernández, J.A. Revisiting the Spectroscopy of Xanthine Derivatives: Theobromine and Theophylline. Phys. Chem. Chem. Phys. 2019, 21, 26430–26437. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrese, C.; Camiruaga, A.; Parra-Santamaria, M.; Evangelisti, L.; Melandri, S.; Maris, A.; Usabiaga, I.; Fernandez, J.A. A Competition between Relative Stability and Binding Energy in Caffeine Phenyl-Glucose Aggregates: Implications in Biological Mechanisms. Int. J. Mol. Sci. 2023, 24, 4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054390
Calabrese C, Camiruaga A, Parra-Santamaria M, Evangelisti L, Melandri S, Maris A, Usabiaga I, Fernandez JA. A Competition between Relative Stability and Binding Energy in Caffeine Phenyl-Glucose Aggregates: Implications in Biological Mechanisms. International Journal of Molecular Sciences. 2023; 24(5):4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054390
Chicago/Turabian StyleCalabrese, Camilla, Ander Camiruaga, Maider Parra-Santamaria, Luca Evangelisti, Sonia Melandri, Assimo Maris, Imanol Usabiaga, and José A. Fernandez. 2023. "A Competition between Relative Stability and Binding Energy in Caffeine Phenyl-Glucose Aggregates: Implications in Biological Mechanisms" International Journal of Molecular Sciences 24, no. 5: 4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054390