
PTB Regulates the Metabolic Pathways and Cell Function of Keloid Fibroblasts through Alternative Splicing of PKM
 , and
, and
Abstract
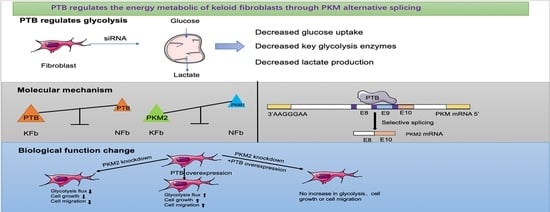
:
1. Introduction
2. Results
2.1. The Expression of PTB Was Abnormally Elevated in Keloid Samples from Patients
2.2. PTB Promotes Glycolysis Flux in KFb
2.3. PTB Promotes Key Glycolysis Enzymes in KFb
2.4. PTB Regulates PKM Splicing in KFb
2.5. PTB Regulates Aerobic Glycolysis through Alternative Splicing of PKM
2.6. PTB Knockdown Downregulated the Key Enzymes of the TCA Cycle
2.7. PKM2 Knockdown Abolished the PTB-Induced Proliferation and Migration of Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Tissue Samples and Cell Culture
4.2. Immunohistochemical (IHC) Staining and Scoring Analyses
4.3. RNA Interference
4.4. Lentivirus Production and Overexpression of PTB and PKM2 in NFb
4.5. RNA Isolation and qRT-PCR
4.6. Western Blotting
4.7. CCK-8 Assay
4.8. Scratch Wound Assay
4.9. Immunofluorescence
4.10. Glucose Uptake and Lactate Production
4.11. RT-PCR and PKM Splicing Assays
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Har-Shai, Y.; Amar, M.; Sabo, E. Intralesional cryotherapy for enhancing the involution of hypertrophic scars and keloids. Plast. Reconstr. Surg. 2003, 111, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R. The Most Current Algorithms for the Treatment and Prevention of Hypertrophic Scars and Keloids: A 2020 Update of the Algorithms Published 10 Years Ago. Plast. Reconstr. Surg. 2022, 149, 79e–94e. [Google Scholar] [CrossRef] [PubMed]
- Trace, A.P.; Enos, C.W.; Mantel, A.; Harvey, V.M. Keloids and Hypertrophic Scars: A Spectrum of Clinical Challenges. Am. J. Clin. Dermatol. 2016, 17, 201–223. [Google Scholar] [CrossRef]
- Mustoe, T.A.; Cooter, R.D.; Gold, M.H.; Hobbs, F.D.; Ramelet, A.A.; Shakespeare, P.G.; Stella, M.; Teot, L.; Wood, F.M.; Ziegler, U.E.; et al. International clinical recommendations on scar management. Plast. Reconstr. Surg. 2002, 110, 560–571. [Google Scholar] [CrossRef]
- Saki, N.; Mokhtari, R.; Nozari, F. Comparing the Efficacy of Intralesional Triamcinolone Acetonide With Verapamil in Treatment of Keloids: A Randomized Controlled Trial. Dermatol. Pract. Concept. 2019, 9, 4–9. [Google Scholar] [CrossRef]
- Arno, A.I.; Gauglitz, G.G.; Barret, J.P.; Jeschke, M.G. Up-to-date approach to manage keloids and hypertrophic scars: A useful guide. Burns 2014, 40, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, M.; Qu, C.; Li, Y.; Tang, Z.; Zhou, Z.; Yu, Z.; Wang, X.; Xin, L.; Shi, T. The Polygenic Map of Keloid Fibroblasts Reveals Fibrosis-Associated Gene Alterations in Inflammation and Immune Responses. Front. Immunol. 2021, 12, 810290. [Google Scholar] [CrossRef]
- He, Y.; Deng, Z.; Alghamdi, M.; Lu, L.; Fear, M.W.; He, L. From genetics to epigenetics: New insights into keloid scarring. Cell Prolif. 2017, 50, 12326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coentro, J.Q.; Pugliese, E.; Hanley, G.; Raghunath, M.; Zeugolis, D.I. Current and upcoming therapies to modulate skin scarring and fibrosis. Adv. Drug. Deliv. Rev. 2019, 146, 37–59. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Liu, W.; Huang, X.; Liang, X.; Zhou, Y.; Li, H.; Yu, Q.; Li, Q. Identification of Key Modules and Hub Genes of Keloids with Weighted Gene Coexpression Network Analysis. Plast. Reconstr. Surg. 2017, 139, 376–390. [Google Scholar] [CrossRef]
- Li, Q.; Qin, Z.; Nie, F.; Bi, H.; Zhao, R.; Pan, B.; Ma, J.; Xie, X. Metabolic reprogramming in keloid fibroblasts: Aerobic glycolysis and a novel therapeutic strategy. Biochem. Biophys. Res. Commun. 2018, 496, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Jiao, H.; Fan, J.; Liu, L.; Tian, J.; Gan, C.; Yang, Z.; Zhang, T.; Chen, Y. Warburg effect in keloids: A unique feature different from other types of scars. Burns 2022, 48, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 7. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 18122755. [Google Scholar] [CrossRef] [Green Version]
- Brix, B.; Mesters, J.R.; Pellerin, L.; Johren, O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J. Neurosci. 2012, 32, 9727–9735. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Vander Heiden, M.G.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Ito, Y.; Otsuki, Y.; Uno, B.; Uchiyama, K.; et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett. 2015, 363, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Nohata, N.; Hanazawa, T.; Enokida, H.; Seki, N. microRNA-1/133a and microRNA-206/133b clusters: Dysregulation and functional roles in human cancers. Oncotarget 2012, 3, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, T.; Yoshikawa, Y.; Inamoto, T.; Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Shinohara, H.; Kumazaki, M.; Tsujino, T.; et al. A Novel Combination RNAi toward Warburg Effect by Replacement with miR-145 and Silencing of PTBP1 Induces Apoptotic Cell Death in Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robida, M.; Sridharan, V.; Morgan, S.; Rao, T.; Singh, R. Drosophila polypyrimidine tract-binding protein is necessary for spermatid individualization. Proc. Natl. Acad. Sci. USA 2010, 107, 12570–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016, 107, 1767–1775. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Chen, X.; Liu, H.; Shao, J.; Xie, R.; Gu, P.; Duan, C. Polypyrimidine Tract-Binding Protein 1 promotes proliferation, migration and invasion in clear-cell renal cell carcinoma by regulating alternative splicing of PKM. Am. J. Cancer Res. 2017, 7, 245–259. [Google Scholar]
- He, X.; Pool, M.; Darcy, K.M.; Lim, S.B.; Auersperg, N.; Coon, J.S.; Beck, W.T. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene 2007, 26, 4961–4968. [Google Scholar] [CrossRef] [Green Version]
- Jiao, H.; Dong, P.; Yan, L.; Yang, Z.; Lv, X.; Li, Q.; Zong, X.; Fan, J.; Fu, X.; Liu, X.; et al. TGF-beta1 Induces Polypyrimidine Tract-Binding Protein to Alter Fibroblasts Proliferation and Fibronectin Deposition in Keloid. Sci. Rep. 2016, 6, 38033. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, P.; Qin, Z.; Yang, X.; Pan, B.; Nie, F.; Bi, H. Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox. Biol. 2021, 38, 101815. [Google Scholar] [CrossRef]
- Dai, S.; Wang, C.; Zhang, C.; Feng, L.; Zhang, W.; Zhou, X.; He, Y.; Xia, X.; Chen, B.; Song, W. PTB: Not just a polypyrimidine tract-binding protein. J. Cell Physiol. 2022, 237, 2357–2373. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Xie, F.; Gong, F.; Yang, Z.; Lv, X.; Li, X.; Jiao, H.; Wang, Q.; Liu, X.; Yan, L.; et al. Suppression of PTBP1 signaling is responsible for mesenchymal stem cell induced invasion of low malignancy cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1552–1565. [Google Scholar] [CrossRef]
- Cho, C.Y.; Chung, S.Y.; Lin, S.; Huang, J.S.; Chen, Y.L.; Jiang, S.S.; Cheng, L.C.; Kuo, T.H.; Lay, J.D.; Yang, Y.Y.; et al. PTBP1-mediated regulation of AXL mRNA stability plays a role in lung tumorigenesis. Sci. Rep. 2019, 9, 16922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Henson, S.M. Divergent mechanisms of metabolic dysfunction drive fibroblast and T-cell senescence. Ageing Res. Rev. 2018, 47, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.S.; Phan, T.T.; Mukhopadhyay, A.; Lim, H.Y.; Halliwell, B.; Wong, K.P. Human skin keloid fibroblasts display bioenergetics of cancer cells. J. Investig. Dermatol. 2008, 128, 702–709. [Google Scholar] [CrossRef]
- Taniguchi, K.; Uchiyama, K.; Akao, Y. PTBP1-targeting microRNAs regulate cancer-specific energy metabolism through the modulation of PKM1/M2 splicing. Cancer Sci. 2021, 112, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Bando, H.; Atsumi, T.; Nishio, T.; Niwa, H.; Mishima, S.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 2005, 11, 5784–5792. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kuranaga, Y.; Sugito, N.; Shinohara, H.; Tsujino, T.; Taniguchi, K.; Komura, K.; Ito, Y.; Soga, T.; Akao, Y. SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Peng, G.; Liu, X.; Zhang, Y.; Han, H.; Liu, Z.R. Pyruvate Kinase M2 Coordinates Metabolism Switch between Glycolysis and Glutaminolysis in Cancer Cells. iScience 2020, 23, 101684. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [Green Version]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Huang, J.Z.; Chen, M.; Chen, D.E.; Gao, X.C.; Zhu, S.; Huang, H.; Hu, M.; Zhu, H.; Yan, G.R. A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol. Cell 2017, 68, 171–184.e6. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Site | Gender | Age (Years) | Period (Months) | Cause |
|---|---|---|---|---|---|
| Normal skin | |||||
| N1 | Eyelid | Female | 25 | ||
| N2 | Eyelid | Female | 27 | ||
| N3 | Eyelid | Female | 19 | ||
| N4 | Eyelid | Female | 29 | ||
| N5 | Eyelid | Female | 18 | ||
| N6 | Eyelid | Female | 23 | ||
| N7 | Eyelid | Female | 28 | ||
| N8 | Abdomen | Female | 36 | ||
| N9 | Eyelid | Female | 50 | ||
| N10 | Hand | Female | 35 | ||
| N11 | Eyelid | Female | 45 | ||
| N12 | Eyelid | Female | 19 | ||
| N13 | Eyelid | Female | 26 | ||
| N14 | Eyelid | Female | 33 | ||
| N15 | Eyelid | Female | 41 | ||
| N16 | Eyelid | Female | 46 | ||
| N17 | Eyelid | Female | 19 | ||
| N18 | Eyelid | Female | 27 | ||
| Keloid | |||||
| * K1 | Perineum | Female | 17 | 14 | Scald |
| * K2 | Shoulder | Female | 21 | 23 | Surgery |
| * K3 | Chest | Male | 26 | 32 | Unknown |
| * K4 | Ear | Male | 24 | 22 | Surgery |
| K5 | Chest | Male | 38 | 12 | Surgery |
| K6 | Neck | Female | 44 | 19 | Surgery |
| K7 | Arm | Male | 41 | 20 | Surgery |
| K8 | Chest | Female | 52 | 13 | Burn |
| K9 | Shoulder | Female | 46 | 16 | Surgery |
| K10 | Shoulder | Female | 29 | 39 | Unknown |
| K11 | Chest | Male | 25 | 13 | Surgery |
| K12 | Ear | Male | 27 | 23 | Unknown |
| K13 | Shoulder | Male | 39 | 30 | Surgery |
| K14 | Chest | Female | 48 | 29 | Surgery |
| K15 | Shoulder | Female | 29 | 19 | Surgery |
| K16 | Arm | Male | 32 | 28 | Burn |
| K17 | Neck | Male | 33 | 120 | Surgery |
| K18 | Shoulder | Male | 45 | 32 | Surgery |
| K19 | Ear | Female | 20 | 14 | Ear piercing |
| K20 | Ear | Female | 39 | 12 | Surgery |
| K21 | Chest | Female | 36 | 36 | Surgery |
| Gene | Primer Sequence (5′ to 3′) |
|---|---|
| PTB F | ACGCACATTCCGTTGCCTTAC |
| PTB R | AACCTGCCTCTACAGCGTCCA |
| GLUT1 F | TGTGGGCArGTGCTTCCAGTA |
| GLUT1 R | CGGCCTTTAGTCTCAGGAACTTTG |
| GLUT3 F | GAGGTGCTGCTCACGTCTC |
| GLUT3 R | GAAACCGTCCGCGTTAAGTT |
| LDHA F | ATGGCAACTCTAAAGGATCAGC |
| LDHA R | CCAACCCCAACAACTGTAATCT |
| HKII F | GAGCCACCACTCACCCTACT |
| HKIIR | CCAGGCATTCGGCAATGTG |
| PFKFB3 F | TTGGCGTCCCCACAAAAGT |
| PFKFB3 R | AGTTGTAGGAGCTGTACTGCTT |
| PKM2 F | TGCCGTGGAGGCCTCCTTCAAGT |
| PKM2 R | GGGGCACGTGGGCGGTATCTG |
| OGDH F | GGCTACGTGTTGACGCCATA |
| OGDH R | CTCAACTTAGCAGCACAAGTCCTTA |
| CS F | GTCTGGCTAACACAGCTGCAGA |
| CS R | CATGGCCATAGCCTGGAACA |
| IDH1 F | AATCAGTGGCGGTTCTGTGGTA |
| IDH1 R | ACTTGGTCGTTGGTGGCATC |
| β-actin F | GTCCACCGCAAATGCTTCTA |
| β-actin R | TGCTGTCACCTTCACCGTTC |
| Antibody | Dilution | Companies |
|---|---|---|
| PTB | 1:1000 (WB) | Abcam, USA |
| PTB | 1:100 (IHC) | Abcam, USA |
| PKM1 | 1:400 | Novus, St. Charles, MO, USA |
| PKM2 | 1:1000 | CST, Boston, MA, USA |
| GLUT1 | 1:1000 | Abcam, USA |
| GLUT3 | 1:1000 | Abcam, USA |
| LDHA | 1:1000 | CST, USA |
| PFKFB3 | 1:1000 | Abcam, USA |
| HKII | 1:1000 | CST, USA |
| OGDH | 1:1000 | CST, USA |
| IDH1 | 1:1000 | Abcam, USA |
| CS | 1:1000 | Abcam, USA |
| β-actin | 1:1000 | ZSGB-BIO, Beijing, China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Han, R.; Yan, Y.; Yang, J.; Dong, G.; Wang, M.; Su, Z.; Jiao, H.; Fan, J. PTB Regulates the Metabolic Pathways and Cell Function of Keloid Fibroblasts through Alternative Splicing of PKM. Int. J. Mol. Sci. 2023, 24, 5162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065162
Huang R, Han R, Yan Y, Yang J, Dong G, Wang M, Su Z, Jiao H, Fan J. PTB Regulates the Metabolic Pathways and Cell Function of Keloid Fibroblasts through Alternative Splicing of PKM. International Journal of Molecular Sciences. 2023; 24(6):5162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065162
Chicago/Turabian StyleHuang, Rong, Rong Han, Yucheng Yan, Jifan Yang, Guoxuan Dong, Miao Wang, Zhiguo Su, Hu Jiao, and Jincai Fan. 2023. "PTB Regulates the Metabolic Pathways and Cell Function of Keloid Fibroblasts through Alternative Splicing of PKM" International Journal of Molecular Sciences 24, no. 6: 5162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065162