
A Proof of Principle Proteomic Study Detects Dystrophin in Human Plasma: Implications in DMD Diagnosis and Clinical Monitoring
 ,
,  and
and
Abstract
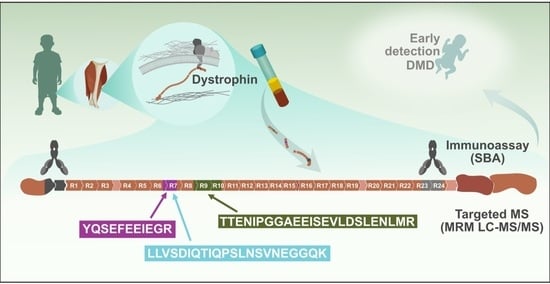
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Suspension Bead Immunoassay
3.2. Targeted Liquid Chromatography Mass Spectrometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Thangarajh, M. The Dystrophinopathies. Continuum 2019, 25, 1619–1639. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.A.; Mahajan, K. Dystrophinopathies; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Thomas, S.; Conway, K.M.; Fapo, O.; Street, N.; Mathews, K.D.; Mann, J.R.; Romitti, P.A.; Soim, A.; Westfield, C.; Fox, D.J.; et al. Time to diagnosis of Duchenne muscular dystrophy remains unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015. Muscle Nerve 2022, 66, 193–197. [Google Scholar] [CrossRef]
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021, 10, 820. [Google Scholar] [CrossRef]
- Chung, J.; Smith, A.L.; Hughes, S.C.; Niizawa, G.; Abdel-Hamid, H.Z.; Naylor, E.W.; Hughes, T.; Clemens, P.R. Twenty-year follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve 2016, 53, 570–578. [Google Scholar] [CrossRef]
- Quinlivan, R. Early diagnosis of Duchenne muscular dystrophy is essential to improve long term outcomes. Arch. Dis. Childhood. 2014, 99, 1061. [Google Scholar] [CrossRef]
- Iragorri, N.; Spackman, E. Assessing the value of screening tools: Reviewing the challenges and opportunities of cost-effectiveness analysis. Public Health Rev. 2018, 39, 17. [Google Scholar] [CrossRef] [Green Version]
- Falzarano, M.S.; Grilli, A.; Zia, S.; Fang, M.; Rossi, R.; Gualandi, F.; Rimessi, P.; El Dani, R.; Fabris, M.; Lu, Z.; et al. RNA-seq in DMD urinary stem cells recognized muscle-related transcription signatures and addressed the identification of atypical mutations by whole-genome sequencing. HGG Adv. 2021, 3, 100054. [Google Scholar] [CrossRef]
- Aravind, S.; Ashley, B.; Mannan, A.; Ganapathy, A.; Ramesh, K.; Ramachandran, A.; Nongthomba, U.; Shastry, A. Targeted sequencing of the DMD locus: A comprehensive diagnostic tool for all mutations. Indian J. Med. Res. 2019, 150, 282–289. [Google Scholar]
- Sun, W.; Zheng, W.; Simeonov, A. Drug discovery and development for rare genetic disorders. Am. J. Med. Genet. A 2017, 173, 2307–2322. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Moore, S.A.; Mathews, K.D. Dystrophinopathy muscle biopsies in the genetic testing ERA: One center’s data. Muscle Nerve 2018, 58, 149–153. [Google Scholar] [CrossRef]
- Strandberg, K.; Ayoglu, B.; Roos, A.; Reza, M.; Niks, E.; Signorelli, M.; Fasterius, E.; Pontén, F.; Lochmüller, H.; Domingos, J.; et al. Blood-derived biomarkers correlate with clinical progression in Duchenne muscular dystrophy. J. Neuromuscul. Dis. 2020, 7, 231–246. [Google Scholar] [CrossRef]
- Meng, Q.; Zhang, J.; Zhong, J.; Zeng, D.; Lan, D. Novel miRNA Biomarkers for Patients with Duchenne Muscular Dystrophy. Front. Neurol. 2022, 13, 921785. [Google Scholar] [CrossRef]
- UK National Screening Committee. Newborn Screening for Duchenne Muscular Dystrophy. Available online: https://view-health-screening-recommendations.service.gov.uk/review/duchenne-muscular-dystrophy-2016/download-documents/cover_sheet/ (accessed on 13 December 2022).
- de Freitas Nakata, K.C.; da Silva Pereira, P.P.; Salgado Riveros, B. Creatine kinase test diagnostic accuracy in neonatal screening for Duchenne Muscular Dystrophy: A systematic review. Clin. Biochem. 2021, 98, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Seol, H.; Han, M.H.J.; Zhang, A.; Brown, K.J.; Hoffman, E.P. Clinical utility of serum biomarkers in Duchenne muscular dystrophy. Clin Proteom. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Ferlini, A.; McNally, E.M.; Spitali, P.; Sweeney, H.L.; workshop participants. 226th ENMC International Workshop: Towards validated and qualified biomarkers for therapy development for Duchenne muscular dystrophy 20–22 January 2017, Heemskerk, The Netherlands. Neuromuscul. Disord. 2018, 28, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Beekman, C.; Janson, A.A.; Baghat, A.; van Deutekom, J.C.; Datson, N.A. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0195850. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Morgan, J.; Lonkar, P.; Neubert, H.; Owens, J.; Binks, M.; Montolio, M.; Phadke, R.; Datson, N.; Van Deutekom, J.; et al. Report of a TREAT-NMD/World Duchenne Organization Meeting on Dystrophin Quantification Methodology. J. Neuromuscul. Dis. 2019, 6, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [PubMed] [Green Version]
- Alnassar, N.; Borczyk, M.; Tsagkogeorga, G.; Korostynski, M.; Han, N.; Górecki, D.C. Full-length dystrophin is expressed across human tissues and DMD downregulation commonly occurring in tumors coincides with Duchenne-like molecular alterations. biorxiv 2022, 4, 486990. [Google Scholar]
- Nimer, R.M.; Sumaily, K.M.; Almuslat, A.; Abdel Jabar, M.; Sabi, E.M.; Al-Muhaizea, M.A.; Abdel Rahman, A.M. Dystrophin Protein Quantification as a Duchenne Muscular Dystrophy Diagnostic Biomarker in Dried Blood Spots Using Multiple Reaction Monitoring Tandem Mass Spectrometry: A Preliminary Study. Molecules 2022, 27, 3662. [Google Scholar] [CrossRef]
- The Human Protein Atlas. 2023. Available online: https://www.proteinatlas.org/ (accessed on 5 January 2023).
- Sun, S.; Henriksen, K.; Karsdal, M.A.; Armbrecht, G.; Belavý, D.L.; Felsenberg, D.; Rittweger, J.; Wang, Y.; Zheng, Q.; Nedergaard, A.F. Measurement of a MMP-2 degraded Titin fragment in serum reflects changes in muscle turnover induced by atrophy. Exp. Gerontol. 2014, 58, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front. Genet. 2021, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Tebani, A.; Gummesson, A.; Zhong, W.; Koistinen, I.S.; Lakshmikanth, T.; Olsson, L.M. Integration of molecular profiles in a longitudinal wellness profiling cohort. Nat. Commun. 2020, 11, 4487. [Google Scholar] [CrossRef]
- Ayoglu, B.; Chaouch, A.; Lochmüller, H.; Politano, L.; Bertini, E.; Spitali, P. Affinity proteomics within rare diseases: A BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014, 6, 918–936. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Harju, A.K.; Bootorabi, F.; Kuuslahti, M.; Supuran, C.T.; Parkkila, S. Carbonic anhydrase III: A neglected isozyme is stepping into the limelight. J. Enzym. Inhib. Med. Chem. 2013, 28, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Fresnais, M.; Longuespée, R. Targeted liquid chromatography-tandem mass spectrometry analysis of proteins: Basic principles, applications, and perspectives. Proteomics 2021, 21, e2100153. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Zheng, R.; Bayer, F.P.; Wong, C.; Chang, Y.C.; Meng, C. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC-MS/MS. Nat. Commun. 2020, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Crutchfield, C.A.; Thomas, S.N.; Sokoll, L.J.; Chan, D.W. Advances in mass spectrometry-based clinical biomarker discovery. Clin. Proteom. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Doykov, I.; Baldwin, T.; Spiewak, J.; Gilmour, K.C.; Gibbons, J.M.; Pade, C.; Reynolds, C.J.; McKnight, A.; Noursadeghi, M.; Maini, M.K.; et al. Quantitative, multiplexed, targeted proteomics for ascertaining variant specific SARS-CoV-2 antibody response. Cell Rep. Methods 2022, 2, 100279. [Google Scholar] [CrossRef] [PubMed]
- Hällqvist, J.; Lane, D.; Shapanis, A.; Davis, K.; Heywood, W.E.; Doykov, I.; Śpiewak, J.; Ghansah, N.; Keevil, B.; Gupta, P.; et al. Operation Moonshot: Rapid translation of a SARS-CoV-2 targeted peptide immunoaffinity liquid chromatography-tandem mass spectrometry test from research into routine clinical use. Clin. Chem. Lab. Med. 2022, 61, 302–310. [Google Scholar] [CrossRef]
- Berglund, L.; Björling, E.; Jonasson, K.; Rockberg, J.; Fagerberg, L.; Al-Khalili Szigyarto, C.; Sivertsson, A.; Uhlén, M. A whole-genome bioinformatics approach to selection of antigens for systematic antibody generation. Proteomics 2008, 8, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Paavilainen, L.; Larsson, K.; Odling, J.; Sundberg, M.; Andersson, A.C.; Kampf, C.; Persson, A.; Al-Khalili Szigyarto, C.; Ottosson, J.; et al. Towards a human proteome atlas: High-throughput generation of mono-specific antibodies for tissue profiling. Proteomics 2005, 5, 4327–4337. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Schwenk, J.M.; Gry, M.; Rimini, R.; Uhlen, M.; Nilsson, P. Antibody suspension bead arrays within serum proteomics. J. Proteome Res. 2008, 7, 3168–3179. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Zhang, Y.; Storey, J.D.; Torres, L.C. Sva: Surrogate Variable Analysis. R Package Version 3.42.0. 2021. Available online: https://www.bioconductor.org/packages/devel/bioc/manuals/sva/man/sva.pdf (accessed on 15 November 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org (accessed on 15 November 2022).
- Berglund, L.; Björling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell. Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegel, H.; Steen, J.; Konrad, A.; Nikdin, H.; Pettersson, K.; Stenvall, M.; Tourle, S.; Wrethagen, U.; Xu, L.; Yderlan, L.; et al. High-throughput protein production-lessons from scaling up from 10 to 288 recombinant proteins per week. Biotechnol. J. 2009, 4, 51–57. [Google Scholar] [CrossRef]
- Edfors, F.; Forsström, B.; Vunk, H.; Kotol, D.; Fredolini, C.; Maddalo, G.; Svensson, A.-S.; Boström, T.; Tegel, H.; Nilsson, P.; et al. Screening a Resource of Recombinant Protein Fragments for Targeted Proteomics. J. Proteome Res. 2019, 18, 2706–2718. [Google Scholar] [CrossRef]
- Skyline Targeted Mass Spec Environment. Available online: https://skyline.ms/project/home/software/Skyline/begin.view (accessed on 20 October 2022).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, R.; Johansson, C.; Heywood, W.; Vinette, H.; Jensen, G.; Tegel, H.; Jiménez-Requena, A.; Torelli, S.; Al-Khalili Szigyarto, C.; Ferlini, A. A Proof of Principle Proteomic Study Detects Dystrophin in Human Plasma: Implications in DMD Diagnosis and Clinical Monitoring. Int. J. Mol. Sci. 2023, 24, 5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065215
Rossi R, Johansson C, Heywood W, Vinette H, Jensen G, Tegel H, Jiménez-Requena A, Torelli S, Al-Khalili Szigyarto C, Ferlini A. A Proof of Principle Proteomic Study Detects Dystrophin in Human Plasma: Implications in DMD Diagnosis and Clinical Monitoring. International Journal of Molecular Sciences. 2023; 24(6):5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065215
Chicago/Turabian StyleRossi, Rachele, Camilla Johansson, Wendy Heywood, Heloise Vinette, Gabriella Jensen, Hanna Tegel, Albert Jiménez-Requena, Silvia Torelli, Cristina Al-Khalili Szigyarto, and Alessandra Ferlini. 2023. "A Proof of Principle Proteomic Study Detects Dystrophin in Human Plasma: Implications in DMD Diagnosis and Clinical Monitoring" International Journal of Molecular Sciences 24, no. 6: 5215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065215