
To Predict, Prevent, and Manage Post-Traumatic Stress Disorder (PTSD): A Review of Pathophysiology, Treatment, and Biomarkers
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Epidemiology and Models of PTSD Development
2.1. Epidemiology of PTSD
2.2. Models of Disease Development
2.2.1. Diathesis–Stress Model for PTSD
2.2.2. Biopsychosocial Model
- Biological disturbance alone is insufficient to cause the disease, as disease appearance results from multi-factor interaction.
- Vulnerability is better accounted for by psychological and social factors than by biological changes.
- The effectiveness of biological treatments is influenced by the psychological status of a “placebo effect.”
- Health outcomes are affected by the doctor–patient relationship to a great extent.
2.2.3. Animal Models of PTSD
- (1)
- Even very brief stressors should cause biological or behavioural symptoms of PTSD;
- (2)
- The stressor should be able to produce symptoms in a dose-dependent manner;
- (3)
- Produced biological alterations should persist or become more pronounced over time;
- (4)
- Alterations should have the potential to express biobehavioural changes in both directions;
- (5)
- Interindividual variability in response is present as a function of experience and/or genetics.
3. Pathophysiology
3.1. Brain Circuits
3.2. Neurochemical Factors
3.2.1. Dysregulation of the Noradrenergic System
3.2.2. Dysregulation of Serotonin Signalling
3.2.3. Dopamine
3.2.4. Gamma-Aminobutyric Acid (GABA)
3.2.5. Neuropeptide Y (NPY)
3.2.6. Brain-Derived Neurotropic Factor (BDNF)
3.2.7. Cannabinoid and Opioid Receptors
3.2.8. Oxytocin
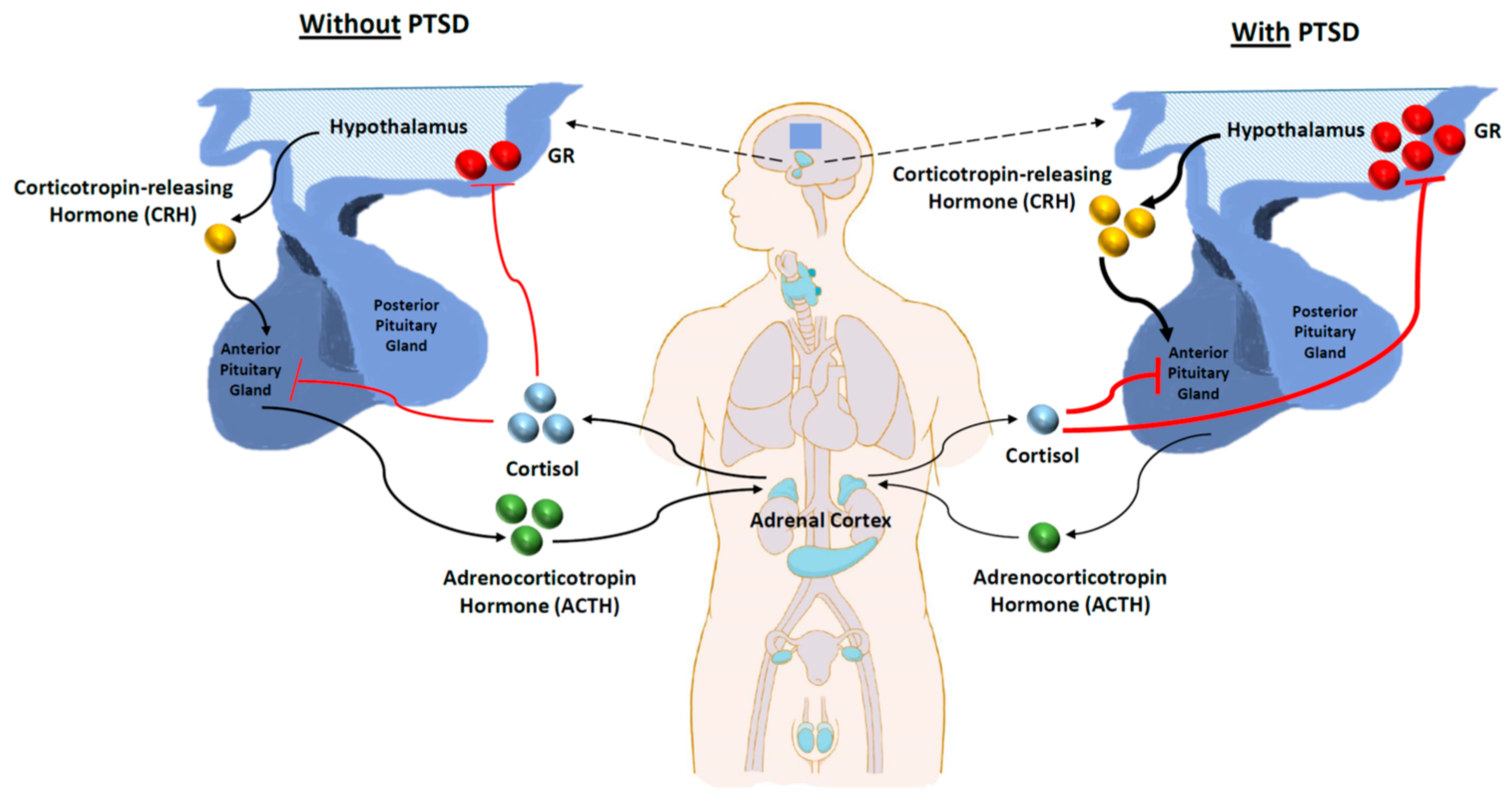
3.3. Dysfunctional HPA Axis
3.4. Conclusive Remarks
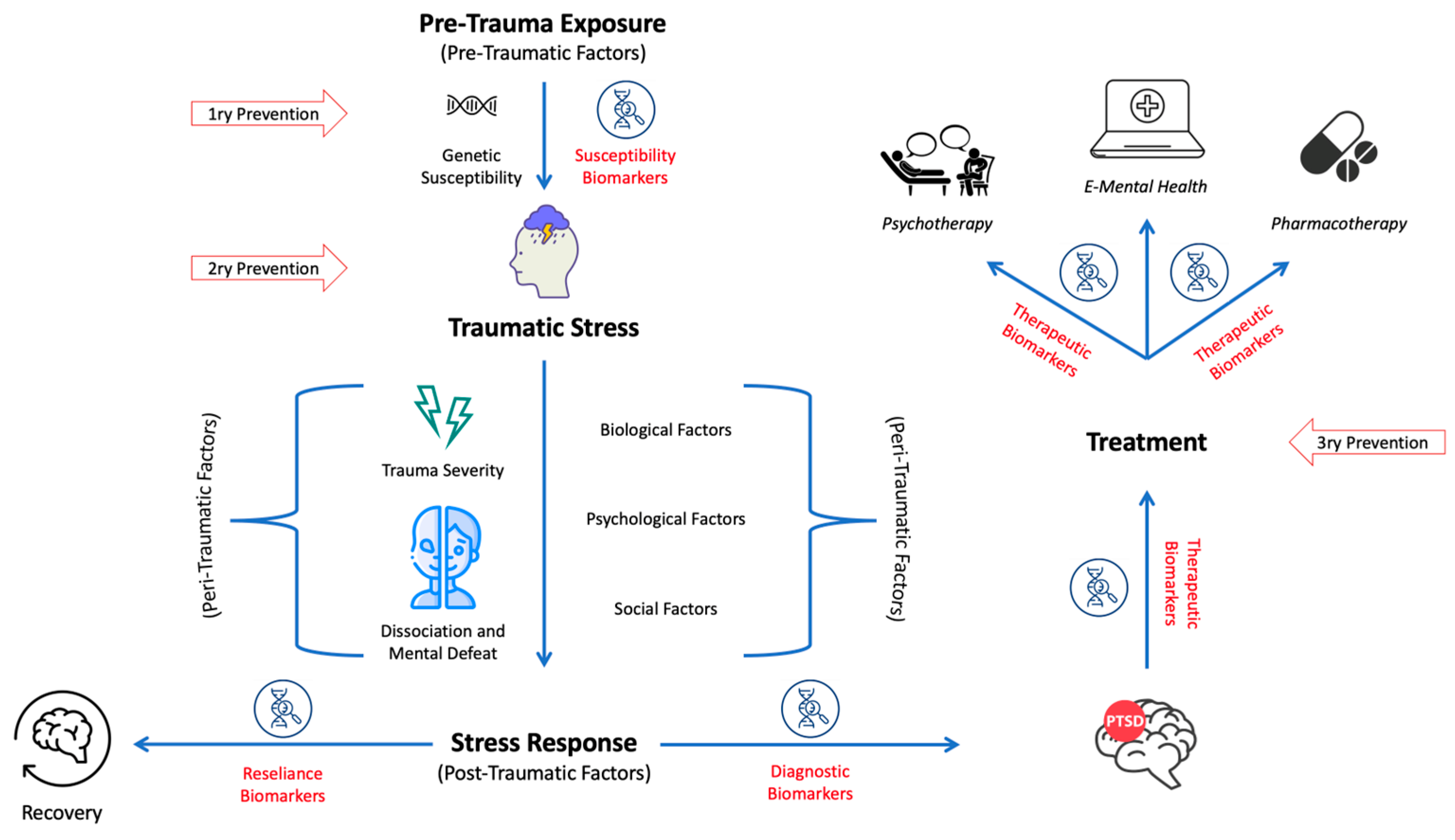
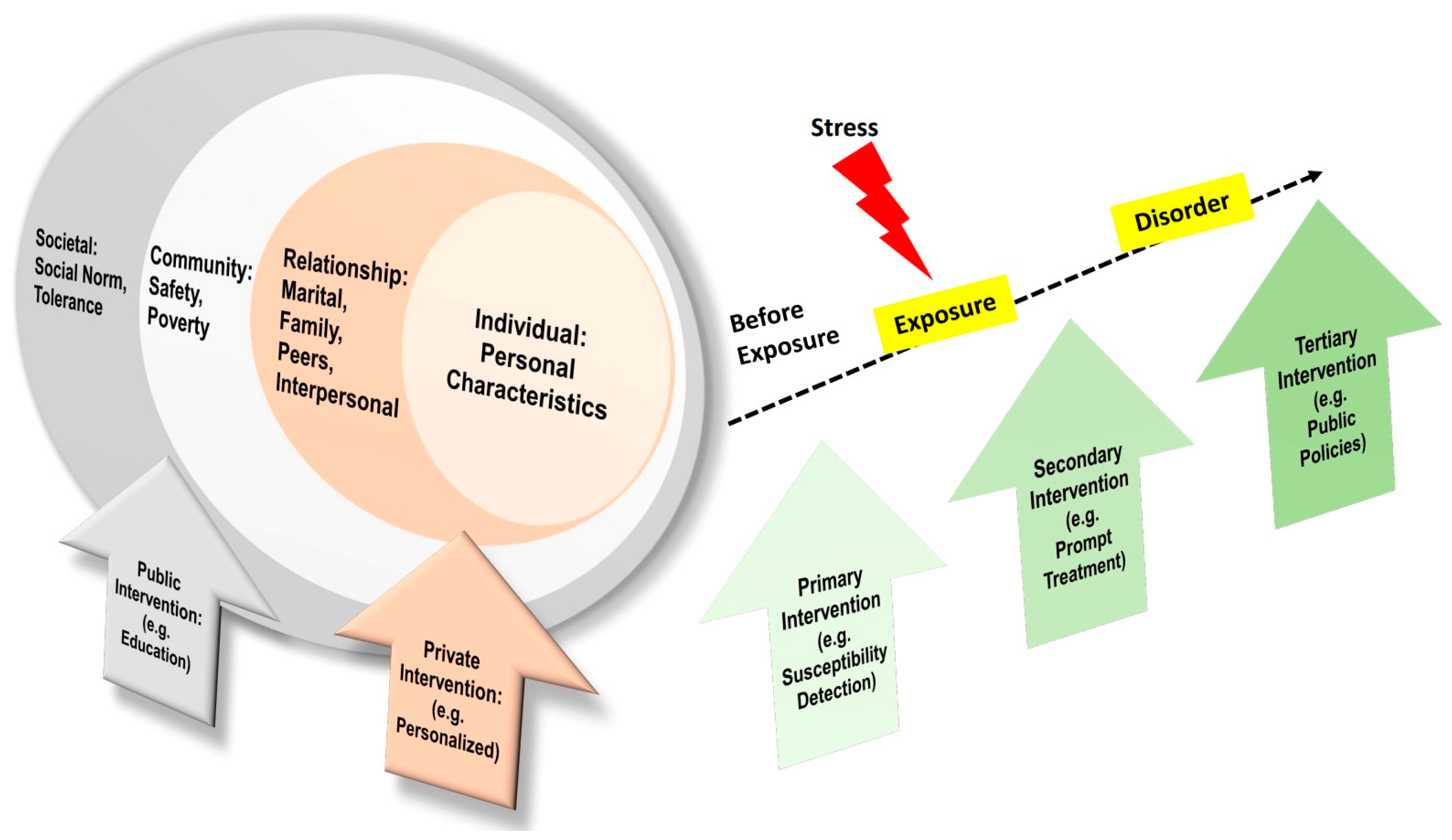
4. Prevention Model of PTSD
The Social-Ecological Model for PTSD Prevention
5. Treatment Modalities for PTSD and E-Mental Health
5.1. Trauma-Based Psychotherapy
5.2. Non-Trauma Focused Psychotherapy
5.3. Pharmacological Therapy
5.4. E-Mental Health and Virtual Reality (VR)
5.5. Conclusive Remarks
6. PTSD Biomarkers
6.1. Susceptibility Biomarkers
6.2. Diagnostic Biomarkers
6.3. Therapeutic Biomarkers
6.4. Conclusive Remarks
7. Limitations and Future Directions
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roehr, B. American Psychiatric Association explains DSM-5. BMJ 2013, 346, f3591. [Google Scholar] [CrossRef] [PubMed]
- Almli, L.M.; Fani, N.; Smith, A.K.; Ressler, K.J. Genetic approaches to understanding post-traumatic stress disorder. Int. J. Neuropsychopharmacol. 2014, 17, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeever, V.M.; Huff, M.E. A diathesis-stress model of posttraumatic stress disorder: Ecological, biological, and residual stress pathways. Rev. Gen. Psychol. 2003, 7, 237–250. [Google Scholar] [CrossRef]
- Sherin, J.E.; Nemeroff, C.B. Post-traumatic stress disorder: The neurobiological impact of psychological trauma. Dialogues Clin. Neurosci. 2011, 13, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Coventry, P.A.; Meader, N.; Melton, H.; Temple, M.; Dale, H.; Wright, K.; Cloitre, M.; Karatzias, T.; Bisson, J.; Roberts, N.P.; et al. Psychological and pharmacological interventions for posttraumatic stress disorder and comorbid mental health problems following complex traumatic events: Systematic review and component network meta-analysis. PLoS Med. 2020, 17, e1003262. [Google Scholar] [CrossRef] [PubMed]
- Hamblen, J.L.; Norman, S.B.; Sonis, J.H.; Phelps, A.J.; Bisson, J.I.; Nunes, V.D.; Megnin-Viggars, O.; Forbes, D.; Riggs, D.S.; Schnurr, P.P. A guide to guidelines for the treatment of posttraumatic stress disorder in adults: An update. Psychotherapy 2019, 56, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo-Martín, M.; Sánchez Sancha, A.; Canto, J.M. Refugee Women with a History of Trauma: Gender Vulnerability in Relation to Post-Traumatic Stress Disorder. Int. J. Environ. Res. Public Health 2021, 18, 4806. [Google Scholar] [CrossRef]
- Hoppen, T.H.; Priebe, S.; Vetter, I.; Morina, N. Global burden of post-traumatic stress disorder and major depression in countries affected by war between 1989 and 2019: A systematic review and meta-analysis. BMJ Glob. Health 2021, 6, e006303. [Google Scholar] [CrossRef]
- Riddle, J.R.; Smith, T.C.; Smith, B.; Corbeil, T.E.; Engel, C.C.; Wells, T.S.; Hoge, C.W.; Adkins, J.; Zamorski, M.; Blazer, D. Millennium Cohort: The 2001–2003 baseline prevalence of mental disorders in the U.S. military. J. Clin. Epidemiol. 2007, 60, 192–201. [Google Scholar] [CrossRef]
- Brewin, C.R.; Andrews, B.; Valentine, J.D. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J. Consult. Clin. Psychol. 2000, 68, 748–766. [Google Scholar] [CrossRef]
- Nasir, B.F.; Black, E.; Toombs, M.; Kisely, S.; Gill, N.; Beccaria, G.; Kondalsamy-Chennakesavan, S.; Nicholson, G. Traumatic life events and risk of post-traumatic stress disorder among the Indigenous population of regional, remote and metropolitan Central-Eastern Australia: A cross-sectional study. BMJ Open 2021, 11, e040875. [Google Scholar] [CrossRef]
- Paradies, Y. Colonisation, racism and indigenous health. J. Popul. Res. 2016, 33, 83–96. [Google Scholar] [CrossRef]
- Bryant-Genevier, J.; Rao, C.Y.; Lopes-Cardozo, B.; Kone, A.; Rose, C.; Thomas, I.; Orquiola, D.; Lynfield, R.; Shah, D.; Freeman, L.; et al. Symptoms of Depression, Anxiety, Post-Traumatic Stress Disorder, and Suicidal Ideation Among State, Tribal, Local, and Territorial Public Health Workers During the COVID-19 Pandemic—United States, March-April 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Mu, E.; Thomas, E.H.X.; Kulkarni, J. Menstrual Cycle in Trauma-Related Disorders: A Mini-Review. Front. Glob. Womens Health 2022, 3, 910220. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.M.; Jerram, M.; Abbs, B.; Whitfield-Gabrieli, S.; Makris, N. Sex differences in stress response circuitry activation dependent on female hormonal cycle. J. Neurosci. 2010, 30, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasiropoulou, C.; Siouti, N.; Mougiakos, T.; Dimitrakopoulos, S. The diathesis-stress model in the emergence of major psychiatric disorders during military service. Psychiatriki 2019, 30, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, D.; Kronish, I.M.; Wasson, L.T.; Giglio, J.F.; Davidson, K.W.; Whang, W. A test of the diathesis-stress model in the emergency department: Who develops PTSD after an acute coronary syndrome? J. Psychiatr. Res. 2014, 53, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Reiss, S. Expectancy model of fear, anxiety, and panic. Clin. Psychol. Rev. 1991, 11, 141–153. [Google Scholar] [CrossRef]
- Alloy, L.B.; Abramson, L.Y.; Safford, S.M.; Gibb, B.E. The cognitive vulnerability to depression (CVD) project: Current findings and future directions. In Cognitive Vulnerability to Emotional Disorders; Routledge: London, UK, 2006; pp. 43–72. [Google Scholar]
- Elwood, L.S.; Mott, J.; Williams, N.L.; Lohr, J.M.; Schroeder, D.A. Attributional style and anxiety sensitivity as maintenance factors of posttraumatic stress symptoms: A prospective examination of a diathesis-stress model. J. Behav. Ther. Exp. Psychiatry 2009, 40, 544–557. [Google Scholar] [CrossRef]
- Engel, G.L. The need for a new medical model: A challenge for biomedicine. Science 1977, 196, 129–136. [Google Scholar] [CrossRef]
- Wade, D.T.; Halligan, P.W. The biopsychosocial model of illness: A model whose time has come. Clin. Rehabil. 2017, 31, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehuda, R.; Antelman, S.M. Criteria for rationally evaluating animal models of posttraumatic stress disorder. Biol. Psychiatry 1993, 33, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Gilpin, N.W.; Edwards, S. Animal models of post-traumatic stress disorder and recent neurobiological insights. Behav. Pharm. 2014, 25, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Van Dijken, H.H.; Van der Heyden, J.A.; Mos, J.; Tilders, F.J. Inescapable footshocks induce progressive and long-lasting behavioural changes in male rats. Physiol. Behav. 1992, 51, 787–794. [Google Scholar] [CrossRef]
- Milad, M.R.; Pitman, R.K.; Ellis, C.B.; Gold, A.L.; Shin, L.M.; Lasko, N.B.; Zeidan, M.A.; Handwerger, K.; Orr, S.P.; Rauch, S.L. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry 2009, 66, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Rau, V.; DeCola, J.P.; Fanselow, M.S. Stress-induced enhancement of fear learning: An animal model of posttraumatic stress disorder. Neurosci. Biobehav. Rev. 2005, 29, 1207–1223. [Google Scholar] [CrossRef]
- Armario, A.; Escorihuela, R.M.; Nadal, R. Long-term neuroendocrine and behavioural effects of a single exposure to stress in adult animals. Neurosci. Biobehav. Rev. 2008, 32, 1121–1135. [Google Scholar] [CrossRef]
- Richter-Levin, G. Acute and long-term behavioral correlates of underwater trauma--potential relevance to stress and post-stress syndromes. Psychiatry Res. 1998, 79, 73–83. [Google Scholar] [CrossRef]
- Yang, R.; Daigle, B.J., Jr.; Muhie, S.Y.; Hammamieh, R.; Jett, M.; Petzold, L.; Doyle, F.J., 3rd. Core modular blood and brain biomarkers in social defeat mouse model for post traumatic stress disorder. BMC Syst. Biol. 2013, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Imanaka, A.; Morinobu, S.; Toki, S.; Yamawaki, S. Importance of early environment in the development of post-traumatic stress disorder-like behaviors. Behav. Brain Res. 2006, 173, 129–137. [Google Scholar] [CrossRef]
- Kim, H.G.; Harrison, P.A.; Godecker, A.L.; Muzyka, C.N. Posttraumatic stress disorder among women receiving prenatal care at three federally qualified health care centers. Matern. Child Health J. 2014, 18, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, P.R.; Conrad, C.D.; Fleshner, M.; Diamond, D.M. Acute episodes of predator exposure in conjunction with chronic social instability as an animal model of post-traumatic stress disorder. Stress 2008, 11, 259–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrechet-Souza, L.; Gilpin, N.W. The predator odor avoidance model of post-traumatic stress disorder in rats. Behav. Pharm. 2019, 30, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A.; Liberzon, I.; Marmar, C. Post-Traumatic Stress Disorder. N. Engl. J. Med. 2017, 376, 2459–2469. [Google Scholar] [CrossRef]
- Cardenas, V.A.; Samuelson, K.; Lenoci, M.; Studholme, C.; Neylan, T.C.; Marmar, C.R.; Schuff, N.; Weiner, M.W. Changes in brain anatomy during the course of posttraumatic stress disorder. Psychiatry Res. 2011, 193, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremner, J.D. Neuroimaging in posttraumatic stress disorder and other stress-related disorders. Neuroimaging Clin. N. Am. 2007, 17, 523–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Ke, J.; Qi, R.; Xu, Q.; Zhong, Y.; Liu, T.; Li, J.; Zhang, L.; Lu, G. Increased Inhibition of the Amygdala by the mPFC may Reflect a Resilience Factor in Post-traumatic Stress Disorder: A Resting-State fMRI Granger Causality Analysis. Front. Psychiatry 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennis, M.; van Rooij, S.J.; van den Heuvel, M.P.; Kahn, R.S.; Geuze, E. Functional network topology associated with posttraumatic stress disorder in veterans. Neuroimage Clin. 2016, 10, 302–309. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, S.J.; Kennis, M.; Vink, M.; Geuze, E. Predicting Treatment Outcome in PTSD: A Longitudinal Functional MRI Study on Trauma-Unrelated Emotional Processing. Neuropsychopharmacology 2016, 41, 1156–1165. [Google Scholar] [CrossRef] [Green Version]
- Hughes, V. Stress: The roots of resilience. Nature 2012, 490, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Pitman, R.K.; Rasmusson, A.M.; Koenen, K.C.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Sripada, R.K.; King, A.P.; Welsh, R.C.; Garfinkel, S.N.; Wang, X.; Sripada, C.S.; Liberzon, I. Neural dysregulation in posttraumatic stress disorder: Evidence for disrupted equilibrium between salience and default mode brain networks. Psychosom. Med. 2012, 74, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgomaneri, S.; Battaglia, S.; Garofalo, S.; Tortora, F.; Avenanti, A.; di Pellegrino, G. State-Dependent TMS over Prefrontal Cortex Disrupts Fear-Memory Reconsolidation and Prevents the Return of Fear. Curr. Biol. 2020, 30, 3672–3679. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. Stopping in (e)motion: Reactive action inhibition when facing valence-independent emotional stimuli. Front. Behav. Neurosci. 2022, 16, 998714. [Google Scholar] [CrossRef]
- Naegeli, C.; Zeffiro, T.; Piccirelli, M.; Jaillard, A.; Weilenmann, A.; Hassanpour, K.; Schick, M.; Rufer, M.; Orr, S.P.; Mueller-Pfeiffer, C. Locus Coeruleus Activity Mediates Hyperresponsiveness in Posttraumatic Stress Disorder. Biol. Psychiatry 2018, 83, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Neumeister, A. Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Res. 2009, 1293, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Liu, A. Catecholamines in Post-traumatic Stress Disorder: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 450. [Google Scholar] [CrossRef]
- Hendrickson, R.C.; Raskind, M.A.; Millard, S.P.; Sikkema, C.; Terry, G.E.; Pagulayan, K.F.; Li, G.; Peskind, E.R. Evidence for altered brain reactivity to norepinephrine in Veterans with a history of traumatic stress. Neurobiol. Stress 2018, 8, 103–111. [Google Scholar] [CrossRef]
- Hendrickson, R.C.; Raskind, M.A. Noradrenergic dysregulation in the pathophysiology of PTSD. Exp. Neurol. 2016, 284, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Keifer, O.P., Jr.; Hurt, R.C.; Ressler, K.J.; Marvar, P.J. The Physiology of Fear: Reconceptualizing the Role of the Central Amygdala in Fear Learning. Physiology 2015, 30, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Giustino, T.F.; Maren, S. Noradrenergic Modulation of Fear Conditioning and Extinction. Front. Behav. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, S.M. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications; Cambridge University Press: Cambridge, UK, 2021. [Google Scholar]
- Brown, R.E.; Basheer, R.; McKenna, J.T.; Strecker, R.E.; McCarley, R.W. Control of sleep and wakefulness. Physiol. Rev. 2012, 92, 1087–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, T.A.; Knorr, B.R.; Pigeon, W.R.; Leiter, J.C.; Akay, M. Heart rate variability during sleep and the early development of posttraumatic stress disorder. Biol. Psychiatry 2004, 55, 953–956. [Google Scholar] [CrossRef]
- Young, S.N. How to increase serotonin in the human brain without drugs. J. Psychiatry Neurosci. 2007, 32, 394–399. [Google Scholar] [PubMed]
- Bailey, C.R.; Cordell, E.; Sobin, S.M.; Neumeister, A. Recent progress in understanding the pathophysiology of post-traumatic stress disorder: Implications for targeted pharmacological treatment. CNS Drugs 2013, 27, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Règue, M.; Poilbout, C.; Martin, V.; Franc, B.; Lanfumey, L.; Mongeau, R. Increased 5-HT2C receptor editing predisposes to PTSD-like behaviors and alters BDNF and cytokines signaling. Transl. Psychiatry 2019, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Browne, C.J.; Ji, X.; Higgins, G.A.; Fletcher, P.J.; Harvey-Lewis, C. Pharmacological Modulation of 5-HT(2C) Receptor Activity Produces Bidirectional Changes in Locomotor Activity, Responding for a Conditioned Reinforcer, and Mesolimbic DA Release in C57BL/6 Mice. Neuropsychopharmacology 2017, 42, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Nebuka, M.; Ohmura, Y.; Izawa, S.; Bouchekioua, Y.; Nishitani, N.; Yoshida, T.; Yoshioka, M. Behavioral characteristics of 5-HT(2C) receptor knockout mice: Locomotor activity, anxiety-, and fear memory-related behaviors. Behav. Brain Res. 2020, 379, 112394. [Google Scholar] [CrossRef]
- Solati, J.; Salari, A.A.; Bakhtiari, A. 5HT(1A) and 5HT(1B) receptors of medial prefrontal cortex modulate anxiogenic-like behaviors in rats. Neurosci. Lett. 2011, 504, 325–329. [Google Scholar] [CrossRef]
- Xiang, M.; Jiang, Y.; Hu, Z.; Yang, Y.; Du, X.; Botchway, B.O.; Fang, M. Serotonin receptors 2A and 1A modulate anxiety-like behavior in post-traumatic stress disordered mice. Am. J. Transl. Res. 2019, 11, 2288–2303. [Google Scholar]
- Sullivan, G.M.; Ogden, R.T.; Huang, Y.Y.; Oquendo, M.A.; Mann, J.J.; Parsey, R.V. Higher in vivo serotonin-1a binding in posttraumatic stress disorder: A PET study with [11C]WAY-100635. Depress. Anxiety 2013, 30, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Bremner, J.D. Posttraumatic Stress Disorder: From Neurobiology to Treatment; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12, 1179069518779829. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.; Reed, M.B.; Pichler, V.; Michenthaler, P.; Rischka, L.; Godbersen, G.M.; Wadsak, W.; Hacker, M.; Lanzenberger, R. Functional dynamics of dopamine synthesis during monetary reward and punishment processing. J. Cereb. Blood Flow Metab. 2021, 41, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Tejada, A.; Neisewander, J.; Katsanos, C.S. Regulation of Voluntary Physical Activity Behavior: A Review of Evidence Involving Dopaminergic Pathways in the Brain. Brain Sci. 2022, 12, 333. [Google Scholar] [CrossRef]
- Comings, D.E.; Muhleman, D.; Gysin, R. Dopamine D2 receptor (DRD2) gene and susceptibility to posttraumatic stress disorder: A study and replication. Biol. Psychiatry 1996, 40, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Gelernter, J.; Southwick, S.; Goodson, S.; Morgan, A.; Nagy, L.; Charney, D.S. No association between D2 dopamine receptor (DRD2) “A” system alleles, or DRD2 haplotypes, and posttraumatic stress disorder. Biol. Psychiatry 1999, 45, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Lawford, B.R.; Noble, E.P.; Kann, B.; Wilkie, A.; Ritchie, T.; Arnold, L.; Shadforth, S. Harmful drinking in military veterans with post-traumatic stress disorder: Association with the D2 dopamine receptor A1 allele. Alcohol Alcohol. 2002, 37, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermetten, E.; Baker, D.G.; Jetly, R.; McFarlane, A.C. Concerns over divergent approaches in the diagnostics of posttraumatic stress disorder. Psychiatr. Ann. 2016, 46, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Mustapic, M.; Maihofer, A.X.; Mahata, M.; Chen, Y.; Baker, D.G.; O’Connor, D.T.; Nievergelt, C.M. The catecholamine biosynthetic enzyme dopamine β-hydroxylase (DBH): First genome-wide search positions trait-determining variants acting additively in the proximal promoter. Hum. Mol. Genet. 2014, 23, 6375–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.C.; Koenen, K.C.; Galea, S.; Aiello, A.E.; Soliven, R.; Wildman, D.E.; Uddin, M. Molecular variation at the SLC6A3 locus predicts lifetime risk of PTSD in the Detroit Neighborhood Health Study. PLoS ONE 2012, 7, e39184. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.S.; Jovanovic, T. Role of social cognition in post-traumatic stress disorder: A review and meta-analysis. Genes Brain Behav. 2019, 18, e12518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arditte Hall, K.A.; DeLane, S.E.; Anderson, G.M.; Lago, T.R.; Shor, R.; Wang, W.; Rasmusson, A.M.; Pineles, S.L. Plasma gamma-aminobutyric acid (GABA) levels and posttraumatic stress disorder symptoms in trauma-exposed women: A preliminary report. Psychopharmacology 2021, 238, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Vaiva, G.; Boss, V.; Ducrocq, F.; Fontaine, M.; Devos, P.; Brunet, A.; Laffargue, P.; Goudemand, M.; Thomas, P. Relationship between posttrauma GABA plasma levels and PTSD at 1-year follow-up. Am. J. Psychiatry 2006, 163, 1446–1448. [Google Scholar] [CrossRef] [PubMed]
- Geuze, E.; van Berckel, B.N.; Lammertsma, A.A.; Boellaard, R.; de Kloet, C.S.; Vermetten, E.; Westenberg, H.G. Reduced GABAA benzodiazepine receptor binding in veterans with post-traumatic stress disorder. Mol. Psychiatry 2008, 13, 74–83. [Google Scholar] [CrossRef]
- Bremner, J.D.; Innis, R.B.; Southwick, S.M.; Staib, L.; Zoghbi, S.; Charney, D.S. Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am. J. Psychiatry 2000, 157, 1120–1126. [Google Scholar] [CrossRef]
- Sah, R.; Geracioti, T.D. Neuropeptide Y and posttraumatic stress disorder. Mol. Psychiatry 2013, 18, 646–655. [Google Scholar] [CrossRef]
- Reichmann, F.; Holzer, P. Neuropeptide Y: A stressful review. Neuropeptides 2016, 55, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Heilig, M.; Koob, G.F.; Ekman, R.; Britton, K.T. Corticotropin-releasing factor and neuropeptide Y: Role in emotional integration. Trends Neurosci. 1994, 17, 80–85. [Google Scholar] [CrossRef]
- Kautz, M.; Charney, D.S.; Murrough, J.W. Neuropeptide Y, resilience, and PTSD therapeutics. Neurosci. Lett. 2017, 649, 164–169. [Google Scholar] [CrossRef]
- Cohen, H.; Liu, T.; Kozlovsky, N.; Kaplan, Z.; Zohar, J.; Mathé, A.A. The neuropeptide Y (NPY)-ergic system is associated with behavioral resilience to stress exposure in an animal model of post-traumatic stress disorder. Neuropsychopharmacology 2012, 37, 350–363. [Google Scholar] [CrossRef]
- Morgan, C.A., 3rd; Wang, S.; Southwick, S.M.; Rasmusson, A.; Hazlett, G.; Hauger, R.L.; Charney, D.S. Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol. Psychiatry 2000, 47, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Schmeltzer, S.N.; Herman, J.P.; Sah, R. Neuropeptide Y (NPY) and posttraumatic stress disorder (PTSD): A translational update. Exp. Neurol. 2016, 284, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Brand, S.; Yang, R.K. Plasma neuropeptide Y concentrations in combat exposed veterans: Relationship to trauma exposure, recovery from PTSD, and coping. Biol. Psychiatry 2006, 59, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Tasan, R.O.; Verma, D.; Wood, J.; Lach, G.; Hörmer, B.; de Lima, T.C.; Herzog, H.; Sperk, G. The role of Neuropeptide Y in fear conditioning and extinction. Neuropeptides 2016, 55, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.X.; Hu, X.Z. Post-traumatic stress disorder risk and brain-derived neurotrophic factor Val66Met. World J. Psychiatry 2016, 6, 1–6. [Google Scholar] [CrossRef]
- Bountress, K.E.; Bacanu, S.A.; Tomko, R.L.; Korte, K.J.; Hicks, T.; Sheerin, C.; Lind, M.J.; Marraccini, M.; Nugent, N.; Amstadter, A.B. The Effects of a BDNF Val66Met Polymorphism on Posttraumatic Stress Disorder: A Meta-Analysis. Neuropsychobiology 2017, 76, 136–142. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Frodl, T.; Schüle, C.; Schmitt, G.; Born, C.; Baghai, T.; Zill, P.; Bottlender, R.; Rupprecht, R.; Bondy, B.; Reiser, M.; et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch. Gen. Psychiatry 2007, 64, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Benedek, D.M.; Fullerton, C.S.; Forsten, R.D.; Naifeh, J.A.; Li, X.X.; Hu, X.Z.; Li, H.; Jia, M.; Xing, G.Q.; et al. PTSD risk is associated with BDNF Val66Met and BDNF overexpression. Mol. Psychiatry 2014, 19, 8–10. [Google Scholar] [CrossRef]
- Bassir Nia, A.; Bender, R.; Harpaz-Rotem, I. Endocannabinoid System Alterations in Posttraumatic Stress Disorder: A Review of Developmental and Accumulative Effects of Trauma. Chronic Stress 2019, 3, 2470547019864096. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Patel, S.; Carrier, E.J.; Rademacher, D.J.; Ormerod, B.K.; Hillard, C.J.; Gorzalka, B.B. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 2005, 30, 508–515. [Google Scholar] [CrossRef]
- Hill, M.N.; Bierer, L.M.; Makotkine, I.; Golier, J.A.; Galea, S.; McEwen, B.S.; Hillard, C.J.; Yehuda, R. Reductions in circulating endocannabinoid levels in individuals with post-traumatic stress disorder following exposure to the World Trade Center attacks. Psychoneuroendocrinology 2013, 38, 2952–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumeister, A.; Normandin, M.D.; Pietrzak, R.H.; Piomelli, D.; Zheng, M.Q.; Gujarro-Anton, A.; Potenza, M.N.; Bailey, C.R.; Lin, S.F.; Najafzadeh, S.; et al. Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: A positron emission tomography study. Mol. Psychiatry 2013, 18, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, J.; Bakos, N.; Szirmay, M.; Ledent, C.; Freund, T.F. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur. J. Neurosci. 2002, 16, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.; Varga, B.; Ledent, C.; Freund, T.F. CB1 cannabinoid receptors mediate anxiolytic effects: Convergent genetic and pharmacological evidence with CB1-specific agents. Behav. Pharm. 2004, 15, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Viveros, M.P.; Llorente, R.; López-Gallardo, M.; Suarez, J.; Bermúdez-Silva, F.; De la Fuente, M.; Rodriguez de Fonseca, F.; Garcia-Segura, L.M. Sex-dependent alterations in response to maternal deprivation in rats. Psychoneuroendocrinology 2009, 34 (Suppl. S1), S217–S226. [Google Scholar] [CrossRef] [Green Version]
- Suárez, J.; Llorente, R.; Romero-Zerbo, S.Y.; Mateos, B.; Bermúdez-Silva, F.J.; de Fonseca, F.R.; Viveros, M.P. Early maternal deprivation induces gender-dependent changes on the expression of hippocampal CB(1) and CB(2) cannabinoid receptors of neonatal rats. Hippocampus 2009, 19, 623–632. [Google Scholar] [CrossRef]
- Boccia, M.L.; Petrusz, P.; Suzuki, K.; Marson, L.; Pedersen, C.A. Immunohistochemical localization of oxytocin receptors in human brain. Neuroscience 2013, 253, 155–164. [Google Scholar] [CrossRef]
- Cardoso, C.; Kingdon, D.; Ellenbogen, M.A. A meta-analytic review of the impact of intranasal oxytocin administration on cortisol concentrations during laboratory tasks: Moderation by method and mental health. Psychoneuroendocrinology 2014, 49, 161–170. [Google Scholar] [CrossRef]
- Knobloch, H.S.; Charlet, A.; Hoffmann, L.C.; Eliava, M.; Khrulev, S.; Cetin, A.H.; Osten, P.; Schwarz, M.K.; Seeburg, P.H.; Stoop, R.; et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 2012, 73, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; Tasker, J.G. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 2012, 204, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Jowf, G.I.; Snijders, C.; Rutten, B.P.F.; de Nijs, L.; Eijssen, L.M.T. The Molecular Biology of Susceptibility to Post-Traumatic Stress Disorder: Highlights of Epigenetics and Epigenomics. Int. J. Mol. Sci. 2021, 22, 10743. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-traumatic stress disorder. Nat. Rev. Dis. Prim. 2015, 1, 15057. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, C.; Barreto, G.E.; Ávila-Rodriguez, M.; Echeverria, V. Role of neuroinflammation and sex hormones in war-related PTSD. Mol. Cell Endocrinol. 2016, 434, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, B.W.; Wong, A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.; Niemeyer, H.; Engel, S.; Cwik, J.C.; Laufer, S.; Klusmann, H.; Knaevelsrud, C. HPA axis regulation in posttraumatic stress disorder: A meta-analysis focusing on potential moderators. Neurosci. Biobehav. Rev. 2019, 100, 35–57. [Google Scholar] [CrossRef]
- Myers, B.; McKlveen, J.M.; Herman, J.P. Glucocorticoid actions on synapses, circuits, and behavior: Implications for the energetics of stress. Front. Neuroendocr. 2014, 35, 180–196. [Google Scholar] [CrossRef] [Green Version]
- Laryea, G.; Arnett, M.G.; Muglia, L.J. Behavioral Studies and Genetic Alterations in Corticotropin-Releasing Hormone (CRH) Neurocircuitry: Insights into Human Psychiatric Disorders. Behav. Sci. 2012, 2, 135–171. [Google Scholar] [CrossRef]
- Bale, T.L.; Contarino, A.; Smith, G.W.; Chan, R.; Gold, L.H.; Sawchenko, P.E.; Koob, G.F.; Vale, W.W.; Lee, K.F. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat. Genet. 2000, 24, 410–414. [Google Scholar] [CrossRef]
- Bale, T.L.; Picetti, R.; Contarino, A.; Koob, G.F.; Vale, W.W.; Lee, K.F. Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior. J. Neurosci. 2002, 22, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coste, S.C.; Kesterson, R.A.; Heldwein, K.A.; Stevens, S.L.; Heard, A.D.; Hollis, J.H.; Murray, S.E.; Hill, J.K.; Pantely, G.A.; Hohimer, A.R.; et al. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat. Genet. 2000, 24, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Sautter, F.J.; Bissette, G.; Wiley, J.; Manguno-Mire, G.; Schoenbachler, B.; Myers, L.; Johnson, J.E.; Cerbone, A.; Malaspina, D. Corticotropin-releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biol. Psychiatry 2003, 54, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Mercer, K.B.; Bradley, B.; Jovanovic, T.; Mahan, A.; Kerley, K.; Norrholm, S.D.; Kilaru, V.; Smith, A.K.; Myers, A.J.; et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 2011, 470, 492–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somvanshi, P.R.; Mellon, S.H.; Yehuda, R.; Flory, J.D.; Makotkine, I.; Bierer, L.; Marmar, C.; Jett, M.; Doyle, F.J., 3rd. Role of enhanced glucocorticoid receptor sensitivity in inflammation in PTSD: Insights from computational model for circadian-neuroendocrine-immune interactions. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E48–E66. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Lehrner, A.; Yehuda, R. Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol. Metab. Clin. N. Am. 2013, 42, 503–513. [Google Scholar] [CrossRef]
- Morris, M.C.; Compas, B.E.; Garber, J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: A systematic review and meta-analysis. Clin. Psychol. Rev. 2012, 32, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, D.G.; Resnick, H.S.; Milanak, M.E.; Miller, M.W.; Keyes, K.M.; Friedman, M.J. National estimates of exposure to traumatic events and PTSD prevalence using DSM-IV and DSM-5 criteria. J. Trauma. Stress 2013, 26, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Al Jowf, G.I.; Ahmed, Z.T.; An, N.; Reijnders, R.A.; Ambrosino, E.; Rutten, B.P.F.; de Nijs, L.; Eijssen, L.M.T. A Public Health Perspective of Post-Traumatic Stress Disorder. Int. J. Environ. Res. Public Health 2022, 19, 6474. [Google Scholar] [CrossRef]
- Magruder, K.M.; Kassam-Adams, N.; Thoresen, S.; Olff, M. Prevention and public health approaches to trauma and traumatic stress: A rationale and a call to action. Eur. J. Psychotraumatol. 2016, 7, 29715. [Google Scholar] [CrossRef] [Green Version]
- Al-Tamimi, S.A.G.; Leavey, G. Community-Based Interventions for the Treatment and Management of Conflict-Related Trauma in Low-Middle Income, Conflict-Affected Countries: A Realist Review. J. Child Adolesc. Trauma 2021, 15, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Kleber, R.J. Trauma and Public Mental Health: A Focused Review. Front. Psychiatry 2019, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Watson, P. PTSD as a Public Mental Health Priority. Curr. Psychiatry Rep. 2019, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Ursano, R.J.; Bell, C.; Eth, S.; Friedman, M.; Norwood, A.; Pfefferbaum, B.; Pynoos, J.D.; Zatzick, D.F.; Benedek, D.M.; McIntyre, J.S.; et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am. J. Psychiatry 2004, 161, 3–31. [Google Scholar]
- Morland, L.A.; Greene, C.J.; Rosen, C.S.; Kuhn, E.; Hoffman, J.; Sloan, D.M. Telehealth and eHealth interventions for posttraumatic stress disorder. Curr. Opin. Psychol. 2017, 14, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S. Cognitive Behavior Therapy: Basics and Beyond; Guilford Publications: New York, NY, USA, 2020. [Google Scholar]
- Forman-Hoffman, V.; Middleton, J.C.; Feltner, C.; Gaynes, B.N.; Weber, R.P.; Bann, C.; Viswanathan, M.; Lohr, K.N.; Baker, C.; Green, J. AHRQ Comparative Effectiveness Reviews. In Psychological and Pharmacological Treatments for Adults With Posttraumatic Stress Disorder: A Systematic Review Update; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2018. [Google Scholar]
- van Minnen, A.; Harned, M.S.; Zoellner, L.; Mills, K. Examining potential contraindications for prolonged exposure therapy for PTSD. Eur. J. Psychotraumatol. 2012, 3, 18805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.M.; Marx, B.P. Written Exposure Therapy for PTSD: A Brief Treatment Approach for Mental Health Professionals; American Psychological Association: Washington, DC, USA, 2019. [Google Scholar]
- Shapiro, F. Eye movement desensitization and reprocessing (EMDR): Evaluation of controlled PTSD research. J. Behav. Ther. Exp. Psychiatry 1996, 27, 209–218. [Google Scholar] [CrossRef]
- Belsher, B.E.; Beech, E.; Evatt, D.; Smolenski, D.J.; Shea, M.T.; Otto, J.L.; Rosen, C.S.; Schnurr, P.P. Present-centered therapy (PCT) for post-traumatic stress disorder (PTSD) in adults. Cochrane Database Syst. Rev. 2019, 2019, CD012898. [Google Scholar] [CrossRef]
- Markowitz, J.C.; Petkova, E.; Neria, Y.; Van Meter, P.E.; Zhao, Y.; Hembree, E.; Lovell, K.; Biyanova, T.; Marshall, R.D. Is Exposure Necessary? A Randomized Clinical Trial of Interpersonal Psychotherapy for PTSD. Am. J. Psychiatry 2015, 172, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Polusny, M.A.; Erbes, C.R.; Thuras, P.; Moran, A.; Lamberty, G.J.; Collins, R.C.; Rodman, J.L.; Lim, K.O. Mindfulness-Based Stress Reduction for Posttraumatic Stress Disorder Among Veterans: A Randomized Clinical Trial. JAMA 2015, 314, 456–465. [Google Scholar] [CrossRef]
- Davidson, J.; Pearlstein, T.; Londborg, P.; Brady, K.T.; Rothbaum, B.; Bell, J.; Maddock, R.; Hegel, M.T.; Farfel, G. Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: Results of a 28-week double-blind, placebo-controlled study. Am. J. Psychiatry 2001, 158, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.J.; Ipser, J.C.; Seedat, S. Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database Syst. Rev. 2006, 2006, Cd002795. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.M.; Neria, Y. Pharmacotherapy in post-traumatic stress disorder: Evidence from randomized controlled trials. Curr. Opin. Investig. Drugs 2009, 10, 35–45. [Google Scholar] [PubMed]
- MacNamara, A.; Rabinak, C.A.; Kennedy, A.E.; Fitzgerald, D.A.; Liberzon, I.; Stein, M.B.; Phan, K.L. Emotion Regulatory Brain Function and SSRI Treatment in PTSD: Neural Correlates and Predictors of Change. Neuropsychopharmacology 2016, 41, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Walderhaug, E.; Kasserman, S.; Aikins, D.; Vojvoda, D.; Nishimura, C.; Neumeister, A. Effects of duloxetine in treatment-refractory men with posttraumatic stress disorder. Pharmacopsychiatry 2010, 43, 45–49. [Google Scholar] [CrossRef]
- Davidson, J.; Baldwin, D.; Stein, D.J.; Kuper, E.; Benattia, I.; Ahmed, S.; Pedersen, R.; Musgnung, J. Treatment of posttraumatic stress disorder with venlafaxine extended release: A 6-month randomized controlled trial. Arch. Gen. Psychiatry 2006, 63, 1158–1165. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, S.; Rosenheck, R.A. Pharmacotherapy of PTSD in the U.S. Department of Veterans Affairs: Diagnostic- and symptom-guided drug selection. J. Clin. Psychiatry 2008, 69, 959–965. [Google Scholar] [CrossRef]
- Katzman, M.A.; Bleau, P.; Blier, P.; Chokka, P.; Kjernisted, K.; Van Ameringen, M.; Antony, M.M.; Bouchard, S.; Brunet, A.; Flament, M.; et al. Canadian clinical practice guidelines for the management of anxiety, posttraumatic stress and obsessive-compulsive disorders. BMC Psychiatry 2014, 14 (Suppl. 1), S1. [Google Scholar] [CrossRef] [Green Version]
- Krystal, J.H.; Rosenheck, R.A.; Cramer, J.A.; Vessicchio, J.C.; Jones, K.M.; Vertrees, J.E.; Horney, R.A.; Huang, G.D.; Stock, C. Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: A randomized trial. JAMA 2011, 306, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, G.; Hamner, M.B.; Cañive, J.M.; Robert, S.; Calais, L.A.; Durklaski, V.; Zhai, Y.; Qualls, C. Efficacy of Quetiapine Monotherapy in Posttraumatic Stress Disorder: A Randomized, Placebo-Controlled Trial. Am. J. Psychiatry 2016, 173, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
- Carey, P.; Suliman, S.; Ganesan, K.; Seedat, S.; Stein, D.J. Olanzapine monotherapy in posttraumatic stress disorder: Efficacy in a randomized, double-blind, placebo-controlled study. Hum. Psychopharmacol. 2012, 27, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Britnell, S.R.; Jackson, A.D.; Brown, J.N.; Capehart, B.P. Aripiprazole for Post-traumatic Stress Disorder: A Systematic Review. Clin. Neuropharmacol. 2017, 40, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.M.; Whiteside, T.E.; Lorenz, R.A.; Wargo, K.A. Prazosin for the treatment of nightmares related to posttraumatic stress disorder: A review of the literature. Prim. Care Companion CNS Disord. 2012, 14, 27120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenen, S.A.; van Wijk, A.J.; van der Heijden, G.J.; van Westrhenen, R.; de Lange, J.; de Jongh, A. Propranolol for the treatment of anxiety disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2016, 30, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangler, P.T.; West, J.C.; Dempsey, C.L.; Possemato, K.; Bartolanzo, D.; Aliaga, P.; Zarate, C.; Vythilingam, M.; Benedek, D.M. Randomized Controlled Trial of Riluzole Augmentation for Posttraumatic Stress Disorder: Efficacy of a Glutamatergic Modulator for Antidepressant-Resistant Symptoms. J. Clin. Psychiatry 2020, 81, 18364. [Google Scholar] [CrossRef]
- Feder, A.; Costi, S.; Rutter, S.B.; Collins, A.B.; Govindarajulu, U.; Jha, M.K.; Horn, S.R.; Kautz, M.; Corniquel, M.; Collins, K.A.; et al. A Randomized Controlled Trial of Repeated Ketamine Administration for Chronic Posttraumatic Stress Disorder. Am. J. Psychiatry 2021, 178, 193–202. [Google Scholar] [CrossRef]
- Rothbaum, B.O.; Price, M.; Jovanovic, T.; Norrholm, S.D.; Gerardi, M.; Dunlop, B.; Davis, M.; Bradley, B.; Duncan, E.J.; Rizzo, A.; et al. A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am. J. Psychiatry 2014, 171, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Guina, J.; Rossetter, S.R.; De, R.B.; Nahhas, R.W.; Welton, R.S. Benzodiazepines for PTSD: A Systematic Review and Meta-Analysis. J. Psychiatr Pract. 2015, 21, 281–303. [Google Scholar] [CrossRef]
- Davis, L.L.; Davidson, J.R.; Ward, L.C.; Bartolucci, A.; Bowden, C.L.; Petty, F. Divalproex in the treatment of posttraumatic stress disorder: A randomized, double-blind, placebo-controlled trial in a veteran population. J. Clin. Psychopharmacol. 2008, 28, 84–88. [Google Scholar] [CrossRef]
- Hamner, M.B.; Faldowski, R.A.; Robert, S.; Ulmer, H.G.; Horner, M.D.; Lorberbaum, J.P. A preliminary controlled trial of divalproex in posttraumatic stress disorder. Ann. Clin. Psychiatry 2009, 21, 89–94. [Google Scholar]
- Davidson, J.R.; Brady, K.; Mellman, T.A.; Stein, M.B.; Pollack, M.H. The efficacy and tolerability of tiagabine in adult patients with post-traumatic stress disorder. J. Clin. Psychopharmacol. 2007, 27, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.S.; Mari, J.J.; Costa, M.C.; Andreoli, S.B.; Bressan, R.A.; Mello, M.F. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNS Neurosci. Ther. 2011, 17, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Frijling, J.L. Preventing PTSD with oxytocin: Effects of oxytocin administration on fear neurocircuitry and PTSD symptom development in recently trauma-exposed individuals. Eur. J. Psychotraumatol. 2017, 8, 1302652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sack, M.; Spieler, D.; Wizelman, L.; Epple, G.; Stich, J.; Zaba, M.; Schmidt, U. Intranasal oxytocin reduces provoked symptoms in female patients with posttraumatic stress disorder despite exerting sympathomimetic and positive chronotropic effects in a randomized controlled trial. BMC Med. 2017, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.C.; Sippel, L.M.; Wahlquist, A.; Moran-Santa Maria, M.M.; Back, S.E. Augmenting Prolonged Exposure therapy for PTSD with intranasal oxytocin: A randomized, placebo-controlled pilot trial. J. Psychiatr. Res. 2018, 98, 64–69. [Google Scholar] [CrossRef]
- Milton, A.L. Fear not: Recent advances in understanding the neural basis of fear memories and implications for treatment development. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef] [Green Version]
- Debiec, J.; Ledoux, J.E. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 2004, 129, 267–272. [Google Scholar] [CrossRef]
- Frewen, P.; Schmahl, C.; Olff, M. Interdisciplinary approaches to understand traumatic stress as a public health problem. Eur. J. Psychotraumatol. 2017, 8, 1441582. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.; Riper, H.; Olff, M. E-health applications in the field of traumatic stress. Eur. J. Psychotraumatol. 2020, 11, 1762317. [Google Scholar] [CrossRef]
- Rizzo, A.S.; Hartholt, A.; Mozgai, S. From Combat to COVID-19–Managing the Impact of Trauma Using Virtual Reality. J. Technol. Hum. Serv. 2021, 39, 314–347. [Google Scholar] [CrossRef]
- Riper, H.; Andersson, G.; Christensen, H.; Cuijpers, P.; Lange, A.; Eysenbach, G. Theme issue on e-mental health: A growing field in internet research. J. Med. Internet Res. 2010, 12, e74. [Google Scholar] [CrossRef]
- Bongaerts, H.; Voorendonk, E.M.; van Minnen, A.; de Jongh, A. Safety and effectiveness of intensive treatment for complex PTSD delivered via home-based telehealth. Eur. J. Psychotraumatol. 2021, 12, 1860346. [Google Scholar] [CrossRef]
- Wind, T.R.; Rijkeboer, M.; Andersson, G.; Riper, H. The COVID-19 pandemic: The ‘black swan’ for mental health care and a turning point for e-health. Internet Interv. 2020, 20, 100317. [Google Scholar] [CrossRef]
- Group, F.-N.B.W. BEST (Biomarkers, EndpointS, and other Tools) Resource; Food and Drug Administration (US): Silver Spring, MD, USA; National Institutes of Health (US): Bethesda, MD, USA, 2016.
- Stein, D.J.; McLaughlin, K.A.; Koenen, K.C.; Atwoli, L.; Friedman, M.J.; Hill, E.D.; Maercker, A.; Petukhova, M.; Shahly, V.; van Ommeren, M.; et al. DSM-5 and ICD-11 definitions of posttraumatic stress disorder: Investigating “narrow” and “broad” approaches. Depress. Anxiety 2014, 31, 494–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wynn, G.H.; Ursano, R.J. A Clinician’s Guide to PTSD Biomarkers and Their Potential Future Use. Focus 2018, 16, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Kaltwasser, S.F.; Wotjak, C.T. Biomarkers in posttraumatic stress disorder: Overview and implications for future research. Dis. Mrk. 2013, 35, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijders, C.; Pries, L.K.; Sgammeglia, N.; Al Jowf, G.; Youssef, N.A.; de Nijs, L.; Guloksuz, S.; Rutten, B.P.F. Resilience Against Traumatic Stress: Current Developments and Future Directions. Front. Psychiatry 2018, 9, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zuiden, M.; Geuze, E.; Willemen, H.L.; Vermetten, E.; Maas, M.; Heijnen, C.J.; Kavelaars, A. Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment. Am. J. Psychiatry 2011, 168, 89–96. [Google Scholar] [CrossRef]
- van Zuiden, M.; Heijnen, C.J.; Maas, M.; Amarouchi, K.; Vermetten, E.; Geuze, E.; Kavelaars, A. Glucocorticoid sensitivity of leukocytes predicts PTSD, depressive and fatigue symptoms after military deployment: A prospective study. Psychoneuroendocrinology 2012, 37, 1822–1836. [Google Scholar] [CrossRef]
- van Zuiden, M.; Geuze, E.; Willemen, H.L.; Vermetten, E.; Maas, M.; Amarouchi, K.; Kavelaars, A.; Heijnen, C.J. Glucocorticoid receptor pathway components predict posttraumatic stress disorder symptom development: A prospective study. Biol. Psychiatry 2012, 71, 309–316. [Google Scholar] [CrossRef]
- Amstadter, A.B.; Nugent, N.R.; Yang, B.Z.; Miller, A.; Siburian, R.; Moorjani, P.; Haddad, S.; Basu, A.; Fagerness, J.; Saxe, G.; et al. Corticotrophin-releasing hormone type 1 receptor gene (CRHR1) variants predict posttraumatic stress disorder onset and course in pediatric injury patients. Dis. Mrk. 2011, 30, 89–99. [Google Scholar] [CrossRef]
- Shalev, A.Y.; Sahar, T.; Freedman, S.; Peri, T.; Glick, N.; Brandes, D.; Orr, S.P.; Pitman, R.K. A prospective study of heart rate response following trauma and the subsequent development of posttraumatic stress disorder. Arch. Gen. Psychiatry 1998, 55, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Liempt, S.; van Zuiden, M.; Westenberg, H.; Super, A.; Vermetten, E. Impact of impaired sleep on the development of PTSD symptoms in combat veterans: A prospective longitudinal cohort study. Depress. Anxiety 2013, 30, 469–474. [Google Scholar] [CrossRef]
- Hinrichs, R.; van Rooij, S.J.; Michopoulos, V.; Schultebraucks, K.; Winters, S.; Maples-Keller, J.; Rothbaum, A.O.; Stevens, J.S.; Galatzer-Levy, I.; Rothbaum, B.O.; et al. Increased Skin Conductance Response in the Immediate Aftermath of Trauma Predicts PTSD Risk. Chronic Stress 2019, 3, 2470547019844441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.K.; Xu, B.; Mukai, J.; Karayiorgou, M.; Gogos, J.A. The BDNF Val66Met variant affects gene expression through miR-146b. Neurobiol. Dis. 2015, 77, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, B.L.; Whealin, J.M.; Harpaz-Rotem, I.; Duman, R.S.; Krystal, J.H.; Southwick, S.M.; Pietrzak, R.H. BDNF Val66Met polymorphism and posttraumatic stress symptoms in U.S. military veterans: Protective effect of physical exercise. Psychoneuroendocrinology 2019, 100, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. Does BDNF Val66Met Polymorphism Confer Risk for Posttraumatic Stress Disorder? Neuropsychobiology 2015, 71, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Bruenig, D.; Lurie, J.; Morris, C.P.; Harvey, W.; Lawford, B.; Young, R.M.; Voisey, J. A Case-Control Study and Meta-Analysis Reveal BDNF Val66Met Is a Possible Risk Factor for PTSD. Neural Plast. 2016, 2016, 6979435. [Google Scholar] [CrossRef] [Green Version]
- Pole, N. The psychophysiology of posttraumatic stress disorder: A meta-analysis. Psychol. Bull. 2007, 133, 725–746. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, B.I.; Catts, S.V. Trauma, PTSD, and physical health: An epidemiological study of Australian Vietnam veterans. J. Psychosom. Res. 2008, 64, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Hellman, N.; Abelson, J.L.; Rao, U. Cortisol, heart rate, and blood pressure as early markers of PTSD risk: A systematic review and meta-analysis. Clin. Psychol. Rev. 2016, 49, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, R.; Neylan, T.C.; Flory, J.D.; McFarlane, A.C. The use of biomarkers in the military: From theory to practice. Psychoneuroendocrinology 2013, 38, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, B.D.; Suvak, M.; Litz, B.T.; Adler, A.B. Heterogeneity in the course of posttraumatic stress disorder: Trajectories of symptomatology. J. Trauma. Stress 2010, 23, 331–339. [Google Scholar] [CrossRef]
- Michopoulos, V.; Norrholm, S.D.; Jovanovic, T. Diagnostic Biomarkers for Posttraumatic Stress Disorder: Promising Horizons from Translational Neuroscience Research. Biol. Psychiatry 2015, 78, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, N.; Bassil, K.; Al Jowf, G.I.; Steinbusch, H.W.M.; Rothermel, M.; de Nijs, L.; Rutten, B.P.F. Dual-specificity phosphatases in mental and neurological disorders. Prog. Neurobiol. 2021, 198, 101906. [Google Scholar] [CrossRef] [PubMed]
- Hawk, L.W.; Dougall, A.L.; Ursano, R.J.; Baum, A. Urinary catecholamines and cortisol in recent-onset posttraumatic stress disorder after motor vehicle accidents. Psychosom. Med. 2000, 62, 423–434. [Google Scholar] [CrossRef]
- Yehuda, R.; Cai, G.; Golier, J.A.; Sarapas, C.; Galea, S.; Ising, M.; Rein, T.; Schmeidler, J.; Müller-Myhsok, B.; Holsboer, F.; et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol. Psychiatry 2009, 66, 708–711. [Google Scholar] [CrossRef]
- Geuze, E.; van Wingen, G.A.; van Zuiden, M.; Rademaker, A.R.; Vermetten, E.; Kavelaars, A.; Fernández, G.; Heijnen, C.J. Glucocorticoid receptor number predicts increase in amygdala activity after severe stress. Psychoneuroendocrinology 2012, 37, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Logue, M.W.; van Rooij, S.J.H.; Dennis, E.L.; Davis, S.L.; Hayes, J.P.; Stevens, J.S.; Densmore, M.; Haswell, C.C.; Ipser, J.; Koch, S.B.J.; et al. Smaller Hippocampal Volume in Posttraumatic Stress Disorder: A Multisite ENIGMA-PGC Study: Subcortical Volumetry Results From Posttraumatic Stress Disorder Consortia. Biol. Psychiatry 2018, 83, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Snijders, C.; Krauskopf, J.; Pishva, E.; Eijssen, L.; Machiels, B.; Kleinjans, J.; Kenis, G.; van den Hove, D.; Kim, M.O.; Boks, M.P.M.; et al. Circulating Serum MicroRNAs as Potential Diagnostic Biomarkers of Posttraumatic Stress Disorder: A Pilot Study. Front. Genet. 2019, 10, 1042. [Google Scholar] [CrossRef]
- Rasmusson, A.M.; Hauger, R.L.; Morgan, C.A.; Bremner, J.D.; Charney, D.S.; Southwick, S.M. Low baseline and yohimbine-stimulated plasma neuropeptide Y (NPY) levels in combat-related PTSD. Biol. Psychiatry 2000, 47, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A., 3rd; Rasmusson, A.M.; Winters, B.; Hauger, R.L.; Morgan, J.; Hazlett, G.; Southwick, S. Trauma exposure rather than posttraumatic stress disorder is associated with reduced baseline plasma neuropeptide-Y levels. Biol. Psychiatry 2003, 54, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Ekhator, N.N.; Strawn, J.R.; Sallee, F.R.; Baker, D.G.; Horn, P.S.; Geracioti, T.D., Jr. Low cerebrospinal fluid neuropeptide Y concentrations in posttraumatic stress disorder. Biol. Psychiatry 2009, 66, 705–707. [Google Scholar] [CrossRef] [Green Version]
- Sah, R.; Ekhator, N.N.; Jefferson-Wilson, L.; Horn, P.S.; Geracioti, T.D., Jr. Cerebrospinal fluid neuropeptide Y in combat veterans with and without posttraumatic stress disorder. Psychoneuroendocrinology 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojtabavi, H.; Saghazadeh, A.; van den Heuvel, L.; Bucker, J.; Rezaei, N. Peripheral blood levels of brain-derived neurotrophic factor in patients with post-traumatic stress disorder (PTSD): A systematic review and meta-analysis. PLoS ONE 2020, 15, e0241928. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Huber, C.; Novak, B.; Schreckenbach, M.; Schubert, C.F.; Touma, C.; Rutten, B.P.; Schmidt, U. Oxytocin receptor is a potential biomarker of the hyporesponsive HPA axis subtype of PTSD and might be modulated by HPA axis reactivity traits in humans and mice. Psychoneuroendocrinology 2021, 129, 105242. [Google Scholar] [CrossRef] [PubMed]
- Blier, P.; El Mansari, M. The importance of serotonin and noradrenaline in anxiety. Int. J. Psychiatry Clin. Pract. 2007, 11 (Suppl. S2), 16–23. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R. Neuroendocrine aspects of PTSD. Handb. Exp. Pharm. 2005, 169, 371–403. [Google Scholar] [CrossRef]
- Meewisse, M.L.; Reitsma, J.B.; de Vries, G.J.; Gersons, B.P.; Olff, M. Cortisol and post-traumatic stress disorder in adults: Systematic review and meta-analysis. Br. J. Psychiatry 2007, 191, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Hauck, S.; Kapczinski, F.; Roesler, R.; de Moura Silveira, E., Jr.; Magalhães, P.V.; Kruel, L.R.; Schestatsky, S.S.; Ceitlin, L.H. Serum brain-derived neurotrophic factor in patients with trauma psychopathology. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 459–462. [Google Scholar] [CrossRef]
- Balakathiresan, N.S.; Chandran, R.; Bhomia, M.; Jia, M.; Li, H.; Maheshwari, R.K. Serum and amygdala microRNA signatures of posttraumatic stress: Fear correlation and biomarker potential. J. Psychiatr Res. 2014, 57, 65–73. [Google Scholar] [CrossRef]
- Hughes, K.C.; Shin, L.M. Functional neuroimaging studies of post-traumatic stress disorder. Expert Rev. Neurother. 2011, 11, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, M.; Grafman, J. Posttraumatic stress disorder: The role of medial prefrontal cortex and amygdala. Neuroscientist 2009, 15, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Admon, R.; Milad, M.R.; Hendler, T. A causal model of post-traumatic stress disorder: Disentangling predisposed from acquired neural abnormalities. Trends Cogn. Sci. 2013, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Carmassi, C.; Marazziti, D.; Mucci, F.; Della Vecchia, A.; Barberi, F.M.; Baroni, S.; Giannaccini, G.; Palego, L.; Massimetti, G.; Dell’Osso, L. Decreased Plasma Oxytocin Levels in Patients With PTSD. Front. Psychol. 2021, 12, 612338. [Google Scholar] [CrossRef] [PubMed]
- Grasser, L.R.; Saad, B.; Bazzi, C.; Wanna, C.; Abu Suhaiban, H.; Mammo, D.; Jovanovic, T.; Javanbakht, A. Skin conductance response to trauma interview as a candidate biomarker of trauma and related psychopathology in youth resettled as refugees. Eur. J. Psychotraumatol. 2022, 13, 2083375. [Google Scholar] [CrossRef]
- Hinrichs, R.; Michopoulos, V.; Winters, S.; Rothbaum, A.O.; Rothbaum, B.O.; Ressler, K.J.; Jovanovic, T. Mobile assessment of heightened skin conductance in posttraumatic stress disorder. Depress. Anxiety 2017, 34, 502–507. [Google Scholar] [CrossRef]
- Elliott, P.; Biddle, D.; Hawthorne, G.; Forbes, D.; Creamer, M. Patterns of treatment response in chronic posttraumatic stress disorder: An application of latent growth mixture modeling. J. Trauma. Stress 2005, 18, 303–311. [Google Scholar] [CrossRef]
- Stein, N.R.; Dickstein, B.D.; Schuster, J.; Litz, B.T.; Resick, P.A. Trajectories of response to treatment for posttraumatic stress disorder. Behav. Ther. 2012, 43, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Felmingham, K.; Kemp, A.; Williams, L.; Das, P.; Hughes, G.; Peduto, A.; Bryant, R. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol. Sci. 2007, 18, 127–129. [Google Scholar] [CrossRef]
- Pagani, M.; Högberg, G.; Salmaso, D.; Nardo, D.; Sundin, O.; Jonsson, C.; Soares, J.; Aberg-Wistedt, A.; Jacobsson, H.; Larsson, S.A.; et al. Effects of EMDR psychotherapy on 99mTc-HMPAO distribution in occupation-related post-traumatic stress disorder. Nucl. Med. Commun. 2007, 28, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.A.; Felmingham, K.; Kemp, A.; Das, P.; Hughes, G.; Peduto, A.; Williams, L. Amygdala and ventral anterior cingulate activation predicts treatment response to cognitive behaviour therapy for post-traumatic stress disorder. Psychol. Med. 2008, 38, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushtaq, D.; Ali, A.; Margoob, M.A.; Murtaza, I.; Andrade, C. Association between serotonin transporter gene promoter-region polymorphism and 4- and 12-week treatment response to sertraline in posttraumatic stress disorder. J. Affect. Disord. 2012, 136, 955–962. [Google Scholar] [CrossRef]
- Berger, W.; Mehra, A.; Lenoci, M.; Metzler, T.J.; Otte, C.; Tarasovsky, G.; Mellon, S.H.; Wolkowitz, O.M.; Marmar, C.R.; Neylan, T.C. Serum brain-derived neurotrophic factor predicts responses to escitalopram in chronic posttraumatic stress disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1279–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, J.S.; Reddy, R.; Kim, Y.J.; van Rooij, S.J.H.; Ely, T.D.; Hamann, S.; Ressler, K.J.; Jovanovic, T. Episodic memory after trauma exposure: Medial temporal lobe function is positively related to re-experiencing and inversely related to negative affect symptoms. Neuroimage Clin. 2018, 17, 650–658. [Google Scholar] [CrossRef]
- Cutsuridis, V.; Yoshida, M. Editorial: Memory Processes in Medial Temporal Lobe: Experimental, Theoretical and Computational Approaches. Front. Syst. Neurosci. 2017, 11, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.; Booth, S.; Spathis, A.; Mollart, S.; Clow, A. Use of Salivary Diurnal Cortisol as an Outcome Measure in Randomised Controlled Trials: A Systematic Review. Ann. Behav. Med. 2016, 50, 210–236. [Google Scholar] [CrossRef] [Green Version]
- Zaba, M.; Kirmeier, T.; Ionescu, I.A.; Wollweber, B.; Buell, D.R.; Gall-Kleebach, D.J.; Schubert, C.F.; Novak, B.; Huber, C.; Köhler, K.; et al. Identification and characterization of HPA-axis reactivity endophenotypes in a cohort of female PTSD patients. Psychoneuroendocrinology 2015, 55, 102–115. [Google Scholar] [CrossRef]
- Rutten, B.P.F.; Vermetten, E.; Vinkers, C.H.; Ursini, G.; Daskalakis, N.P.; Pishva, E.; de Nijs, L.; Houtepen, L.C.; Eijssen, L.; Jaffe, A.E.; et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol. Psychiatry 2018, 23, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Snijders, C.; Maihofer, A.X.; Ratanatharathorn, A.; Baker, D.G.; Boks, M.P.; Geuze, E.; Jain, S.; Kessler, R.C.; Pishva, E.; Risbrough, V.B.; et al. Longitudinal epigenome-wide association studies of three male military cohorts reveal multiple CpG sites associated with post-traumatic stress disorder. Clin. Epigenet. 2020, 12, 11. [Google Scholar] [CrossRef]
- Logue, M.W.; Miller, M.W.; Wolf, E.J.; Huber, B.R.; Morrison, F.G.; Zhou, Z.; Zheng, Y.; Smith, A.K.; Daskalakis, N.P.; Ratanatharathorn, A.; et al. An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin. Epigenet. 2020, 12, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, D.; Bruenig, D.; Carrillo-Roa, T.; Lawford, B.; Harvey, W.; Morris, C.P.; Smith, A.K.; Binder, E.B.; Young, R.M.; Voisey, J. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr. Scand. 2017, 136, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlim, M.T.; Van Den Eynde, F. Repetitive transcranial magnetic stimulation over the dorsolateral prefrontal cortex for treating posttraumatic stress disorder: An exploratory meta-analysis of randomized, double-blind and sham-controlled trials. Can. J. Psychiatry 2014, 59, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, K.; Andersen, S.B.; Carlsson, J. Neurofeedback Treatment and Posttraumatic Stress Disorder: Effectiveness of Neurofeedback on Posttraumatic Stress Disorder and the Optimal Choice of Protocol. J. Nerv. Ment. Dis. 2016, 204, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, S.; Vancampfort, D.; Steel, Z.; Newby, J.; Ward, P.B.; Stubbs, B. Physical activity in the treatment of Post-traumatic stress disorder: A systematic review and meta-analysis. Psychiatry Res. 2015, 230, 130–136. [Google Scholar] [CrossRef]
- Reznikov, R.; Hamani, C. Posttraumatic Stress Disorder: Perspectives for the Use of Deep Brain Stimulation. Neuromodulation 2017, 20, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Model | Description | Aim |
|---|---|---|
| Physical: | ||
| Foot shock stress (FSS) | A metal-rod floor is used to deliver electrical shocks, which are coupled with non-harmful cues (usually auditory) to elicit post-stress fear recall using sound in novel environments, according to the fear conditioning model | Modelling the response to inescapable stress [25] |
| Single prolonged stress (SPS) | Intends to result in the development of PTSD from a single traumatic experience. Rats are restrained for 2 h, subsequently forced to swim for 20 min, and then 15 min later are subjected to the ether until unconsciousness | Inducing PTSD symptoms by combining multiple, severe, and different stressors [26] |
| Stress-enhanced fear learning (SEFL) | Exposure to repeated foot shocks in one environment produces conditional freezing (used to assess learned fear) in response to cues associated with foot shock in a second environment | Modelling the lasting effects of traumatic stress [27] |
| Restraint stress (RS) | Prolonged restraint between 15 min to 2 h on a wooden board or a plastic tube | Modelling an inescapable severe psychological trauma with chronic behavioural and neurochemical alterations [28] |
| Underwater trauma (UWT) | Being distinct from the forced-swim test, animals are placed in deep water and are forced to swim for 30 s, before being submersed for another 30 s | Modelling an inescapable severe psychological trauma [29] |
| Social: | ||
| Social defeat (SD) | Subjects are categorised as either susceptible or resilient and are exposed to and suppressed by an aggressor animal for several days. Only susceptible subjects will develop behavioural avoidance | Modelling prolonged and chronic stress as a risk of PTSD [30] |
| Early life stress (ELS) | From postnatal days 2–14, new-born mice are separated from their mother 1 h daily | Modelling the ability of childhood trauma to influence the development of PTSD [31] |
| Housing instability (HI) | Animals are frequently paired with different cohorts, after being exposed to their natural predators (e.g., cats) | Modelling the effects of housing instability in PTSD patients [32] |
| Psychological: | ||
| Predator scent stress (PSS) | Animals are confronted with the scents of their natural predators (cat litter, urine, etc.) | Modelling and simulating traumatising events and trauma-related stimulus response in humans [33,34] |
| Susceptibility Biomarker | Findings |
|---|---|
| Number of GR in lymphocytes and monocytes | A higher number pre-trauma of GR is associated with high PTSD symptoms in soldiers after deployment [175]. |
| Sensitivity of T cells to dexamethasone before deployment | High sensitivity pre-trauma is associated with a high amount of PTSD symptoms without comorbid depressive symptoms. Different sensitivity patterns are associated with different symptomatology [176]. |
| mRNA levels of FKBP5 | Low levels after deployment are associated with a high amount of PTSD symptoms [176]. |
| Glucocorticoid-induced leucine zipper mRNA | High levels pre-trauma are associated with a high amount of PTSD symptoms post-deployment [177]. |
| Corticotropin-releasing hormone type 1 receptor gene | Polymorphisms were associated with PTSD development [178]. |
| Heart rate | Increased heart rates in the post-traumatic period were associated with PTSD development [179]. |
| Occurrence of Nightmares | Higher occurrence of nightmares pre-trauma was associated with disease susceptibility in Dutch combat soldiers [180]. |
| Increased skin conductance | Skin conductance response (SCR) within hours of trauma exposure was a predictor of chronic PTSD development [181]. |
| Diagnostic Biomarker | Findings |
|---|---|
| Noradrenaline levels | Increased urinary noradrenaline levels were associated with PTSD development in men [193]. |
| FKBP5 levels | Reduced FKBP5 expression in blood was found in PTSD patients [194]. |
| Amygdala activity | Amygdala over-activation is found in PTSD patients [42,195]. |
| Hippocampus volume | Hippocampal loss is a common anatomical change in patients with PTSD [196]. |
| miR-138-5p overexpression miR-1246 downregulation | Plasma isolated miR-138-5p was significantly overexpressed in subjects with PTSD compared to controls, and miR-1246 was significantly downregulated in subjects with PTSD compared to resilient subjects [197]. |
| Plasma levels of NPY | Plasma baseline levels are lower in individuals with traumatic stress exposure and PTSD [198,199]. |
| CSF levels of NPY | Levels of NPY were lower in combat veterans with PTSD compared to veterans without PTSD and healthy controls [200,201]. |
| Plasma BDNF level | Patients with PTSD have higher plasma levels of BDNF [202]. |
| Oxytocin receptor mRNA levels | Blood mRNA levels of OXTR were lower in patients with hyporeactive HPA axis subtype at baseline, which increased during stress testing [203]. |
| Others | A rise in inflammatory markers, increased startle response, symptoms of hyperarousal, and impaired cognitive function were found in PTSD patients [173,191]. |
| Therapeutic Biomarker | Findings |
|---|---|
| Amygdala and anterior cingulate cortex activity | Successful cognitive behavioural therapy was observed to decrease right amygdala activity while increasing right anterior cingulate cortex activity [217]. |
| Cerebral blood flow to the medial temporal cortex | A normalisation of the difference in cerebral blood flow to the medial temporal cortex after EMDR [218]. |
| Amygdala and ventral anterior cingulate activity | The greater activation of the bilateral amygdala and ventral anterior cingulate was associated with poorer response to CBT [219]. |
| Rostral anterior cingulate (rACC) volume | A larger rostral anterior cingulate (rACC) volume was also found in responders to CBT [219]. |
| LL genotype of serotonin transporter gene promoter (5HTTLPR) | A polymorphism in the LL genotype 5HTTLPR was found to be associated with a better response to sertraline [220]. |
| BDNF levels | Lower serum levels of BDNF were associated with a decrease in PTSD symptoms in chronic patients on escitalopram [221]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Jowf, G.I.; Ahmed, Z.T.; Reijnders, R.A.; de Nijs, L.; Eijssen, L.M.T. To Predict, Prevent, and Manage Post-Traumatic Stress Disorder (PTSD): A Review of Pathophysiology, Treatment, and Biomarkers. Int. J. Mol. Sci. 2023, 24, 5238. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065238
Al Jowf GI, Ahmed ZT, Reijnders RA, de Nijs L, Eijssen LMT. To Predict, Prevent, and Manage Post-Traumatic Stress Disorder (PTSD): A Review of Pathophysiology, Treatment, and Biomarkers. International Journal of Molecular Sciences. 2023; 24(6):5238. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065238
Chicago/Turabian StyleAl Jowf, Ghazi I., Ziyad T. Ahmed, Rick A. Reijnders, Laurence de Nijs, and Lars M. T. Eijssen. 2023. "To Predict, Prevent, and Manage Post-Traumatic Stress Disorder (PTSD): A Review of Pathophysiology, Treatment, and Biomarkers" International Journal of Molecular Sciences 24, no. 6: 5238. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065238