
An Efficient 2D Protocol for Differentiation of iPSCs into Mature Postmitotic Dopaminergic Neurons: Application for Modeling Parkinson’s Disease
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Development of an Efficient and Reproducible 2D Protocol for iPSC Differentiation into Tyrosine Hydroxylase-Positive Neurons

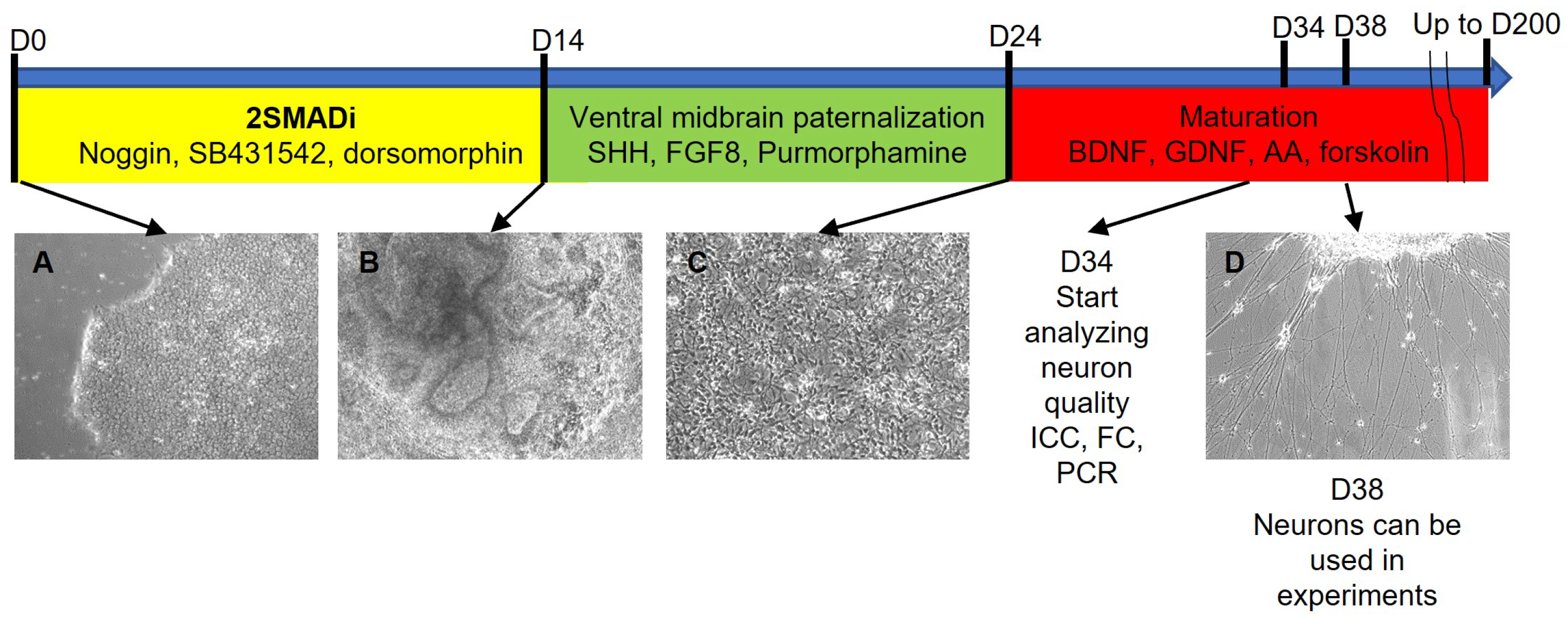{kind=link}
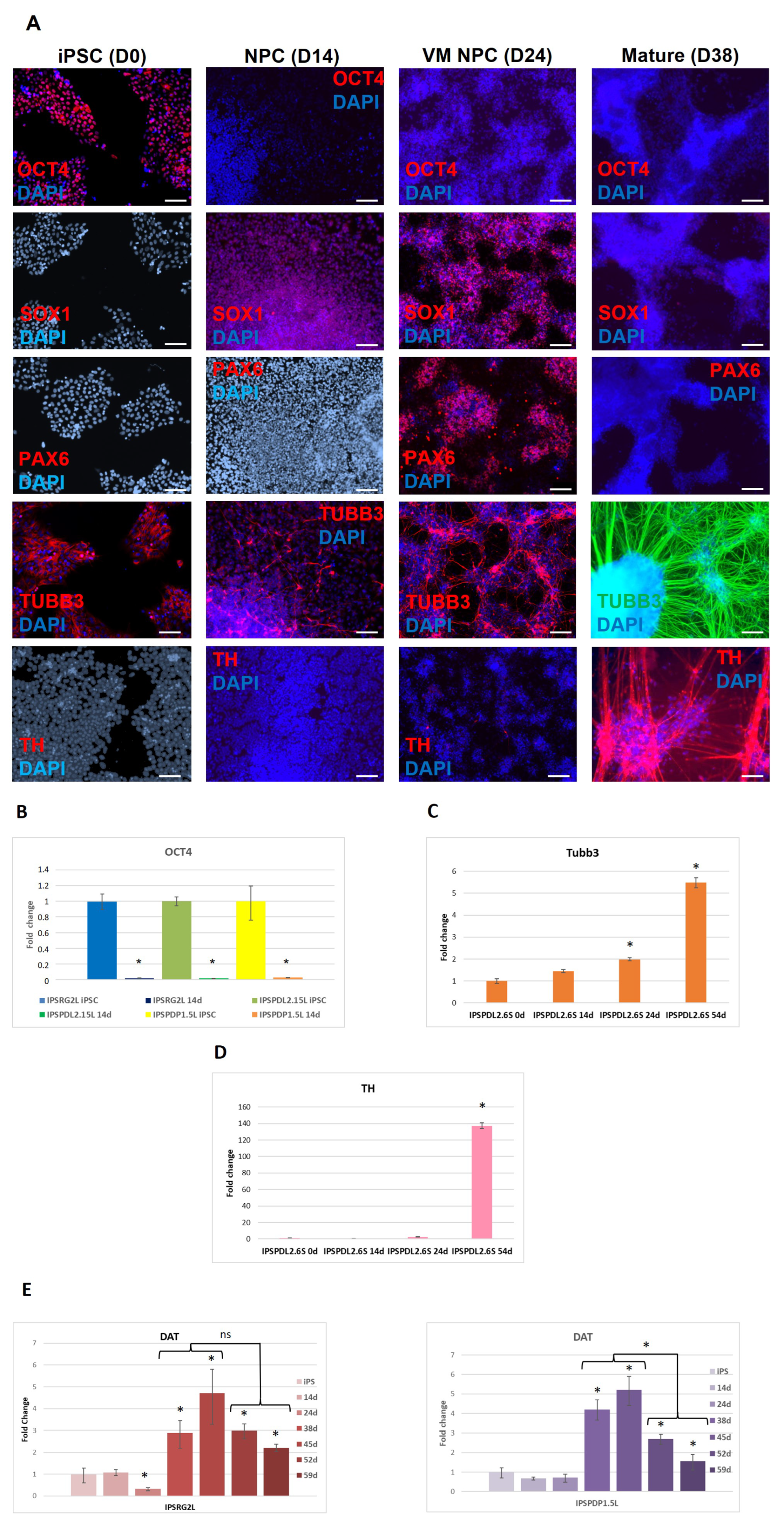{kind=link}
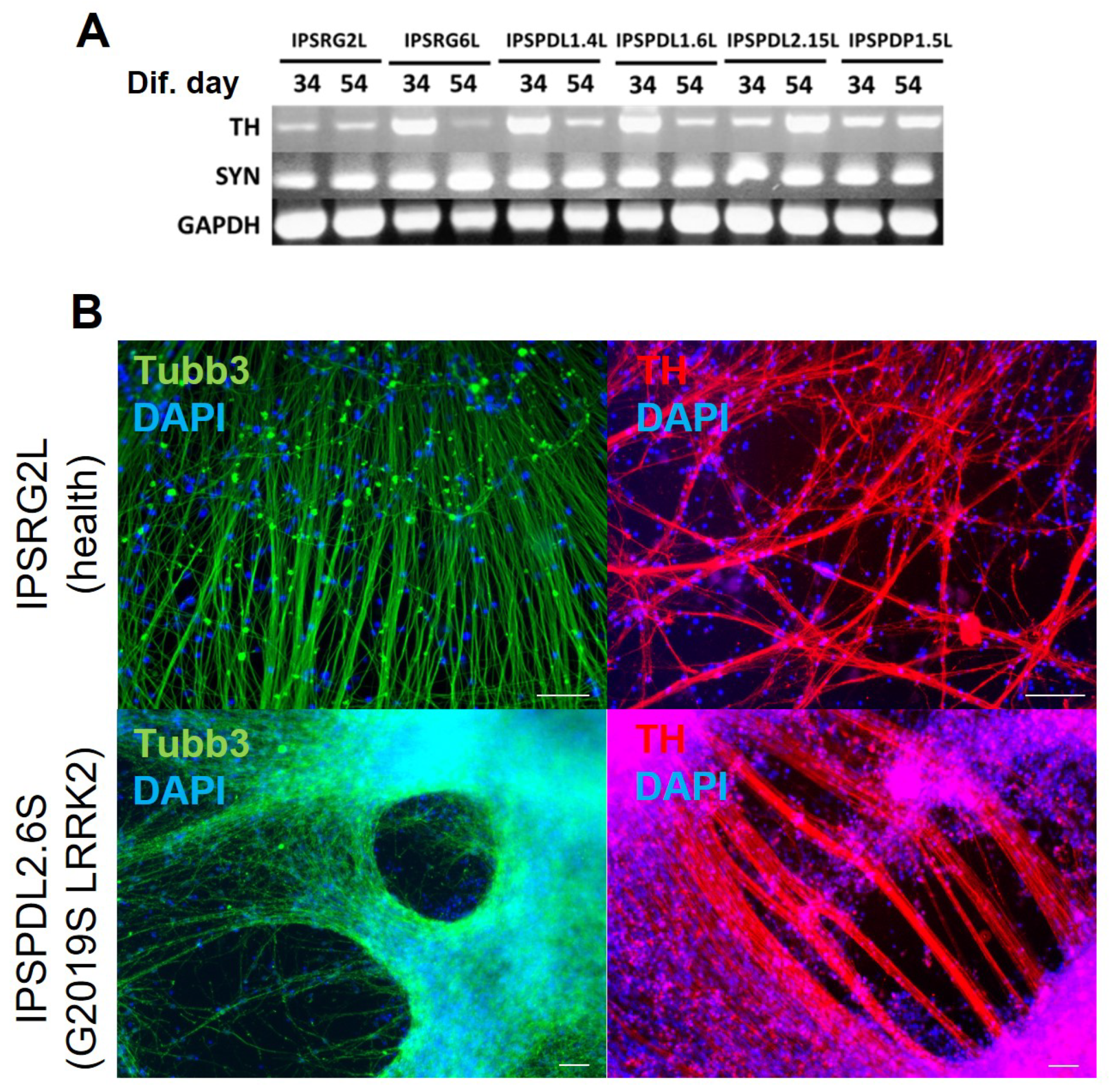{kind=link}
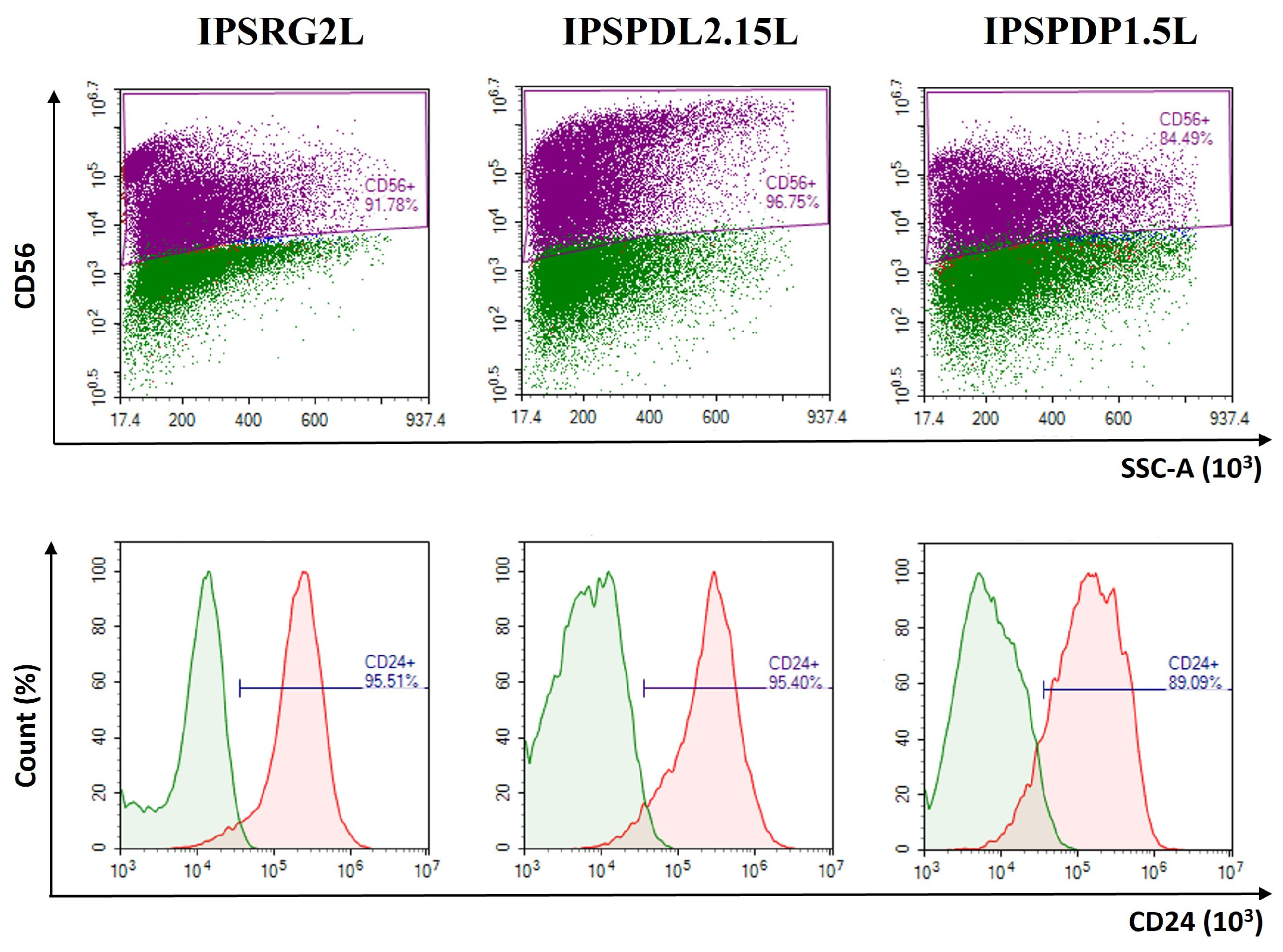{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Biopsy Age | Mutation | Fibroblasts | iPSCs | Method | Reference |
|---|---|---|---|---|---|---|---|
| RG | M | 60 | - | FRG | IPSRG2L | Lenti | [14] |
| IPSRG6L | Lenti | [14] | |||||
| IPSRG4S | Sev | [14] | |||||
| FF | F | 48 | - | FF | IPSFF1S | Sev | [15] |
| PDL1 | M | 60 | G2019S in LRRK2 | FPDL1 | IPSPDL1.4L | Lenti | [14] |
| (PARK8 gene) | IPSPDL1.6L | Lenti | [14] | ||||
| and N370S GBA | IPSPDL1.6S | Sev | [14] | ||||
| PDL2 | M | 60 | G2019S in LRRK2 | FPDL2 | IPSPDL2.15L | Lenti | [14] |
| (PARK8 gene) | IPSPDL2.6S | Sev | [14] | ||||
| PDP1 | M | 65 | deletion 202–203 AG | FPDP1 | IPSPDP1.5L | Lenti | Supplementary Figure S1 [16] |
| in the second exon and splicing mutation in 1 intron (IVS1+1G/A) in PARK2 gene |
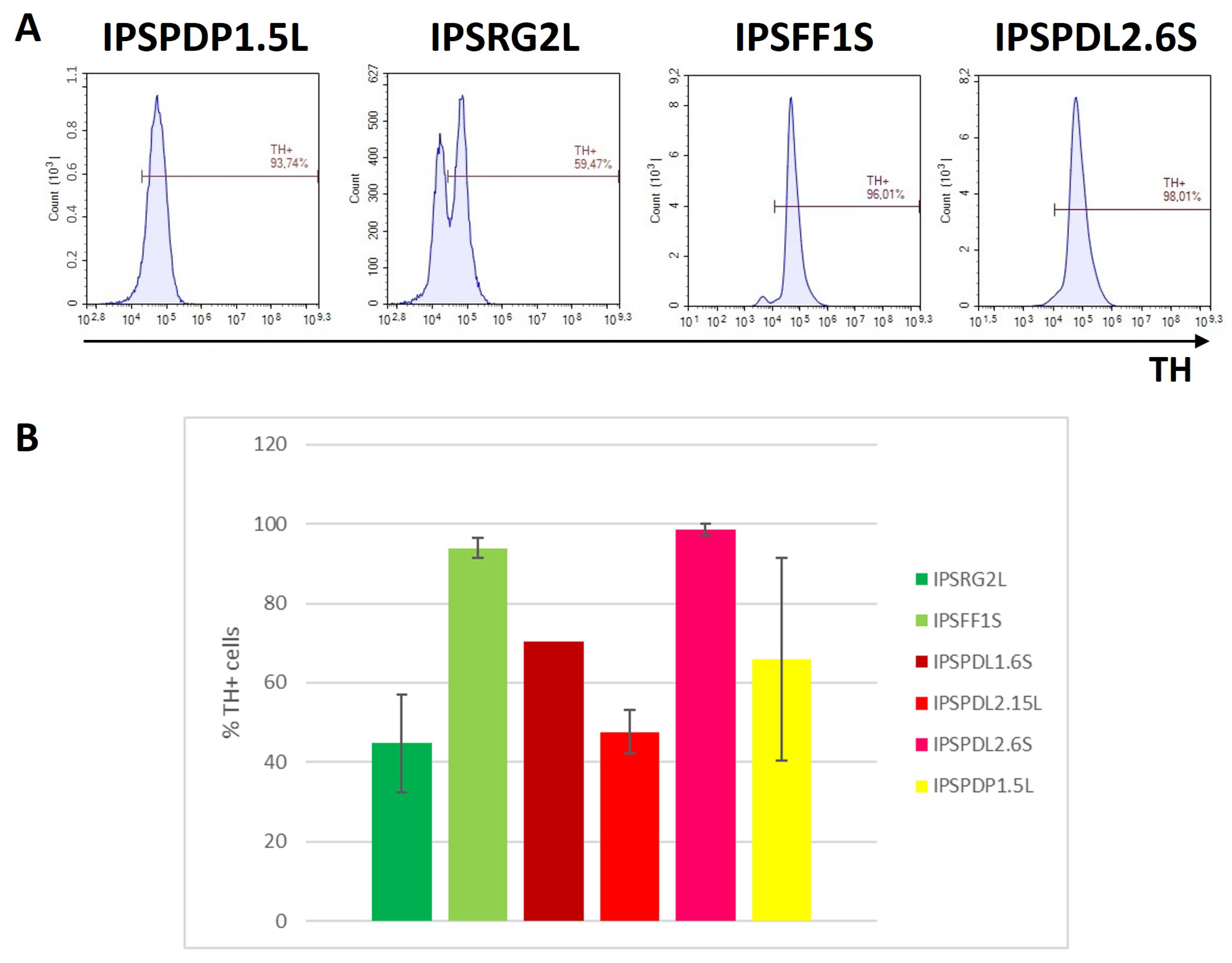
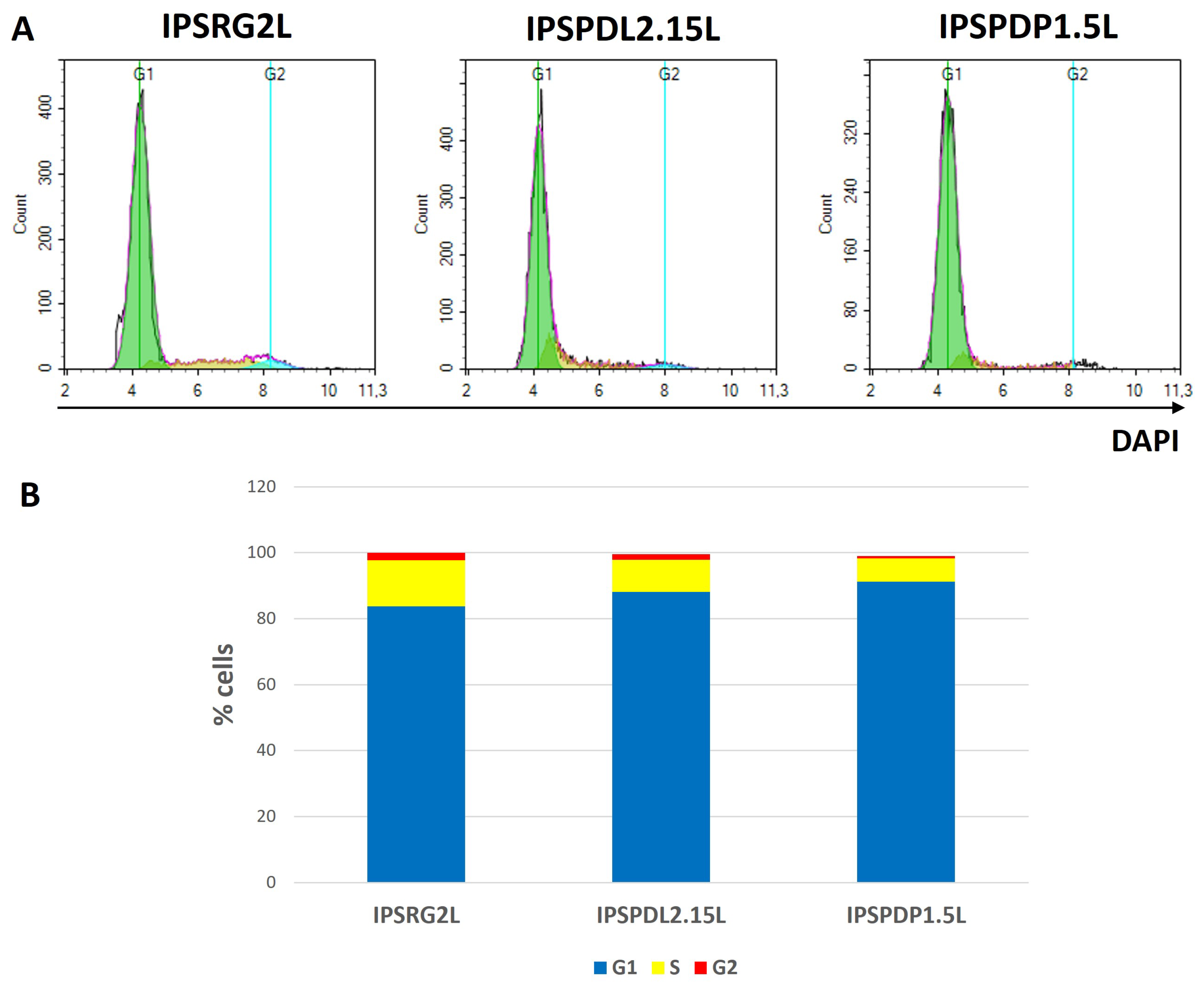
2.2. Determination of Neuronal Differentiation Efficiency Using Flow Cytometry
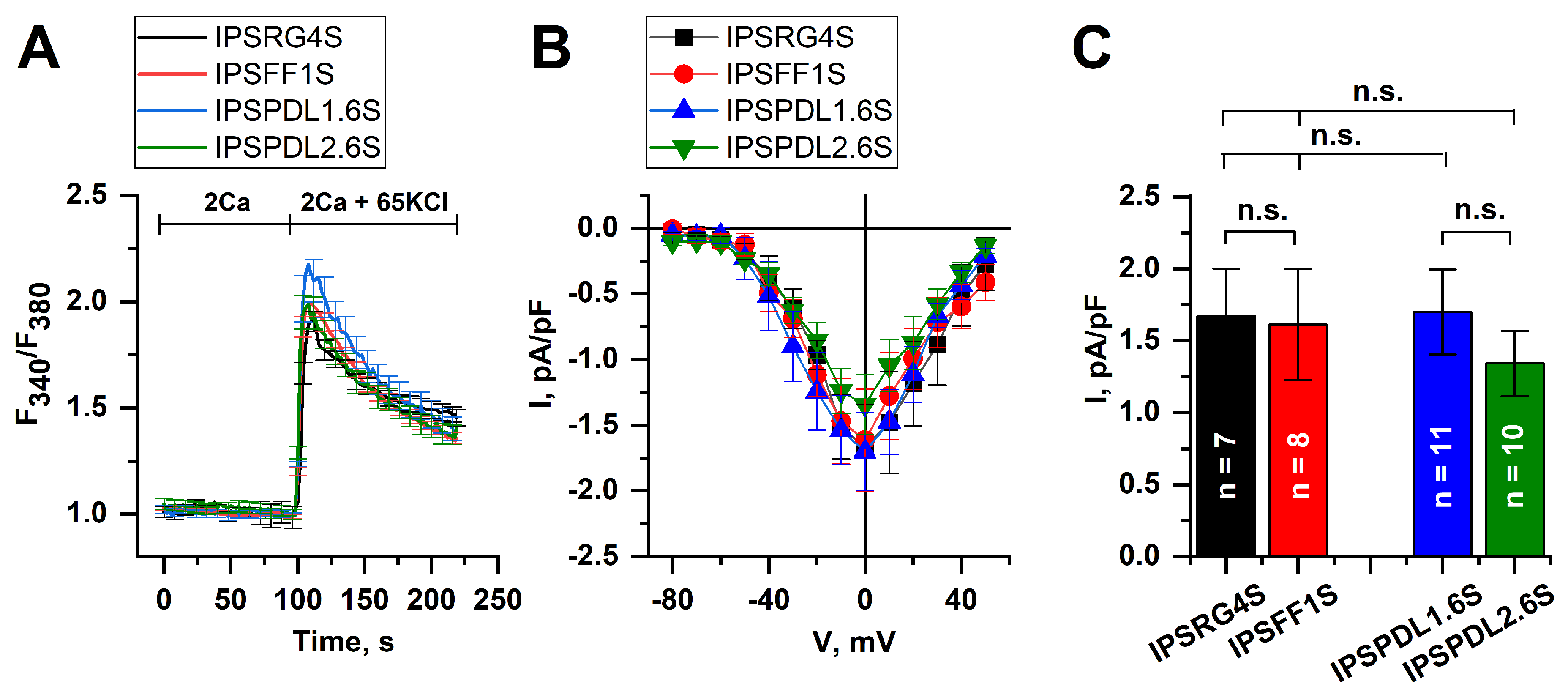
2.3. Analysis of Neuron Functional Activity
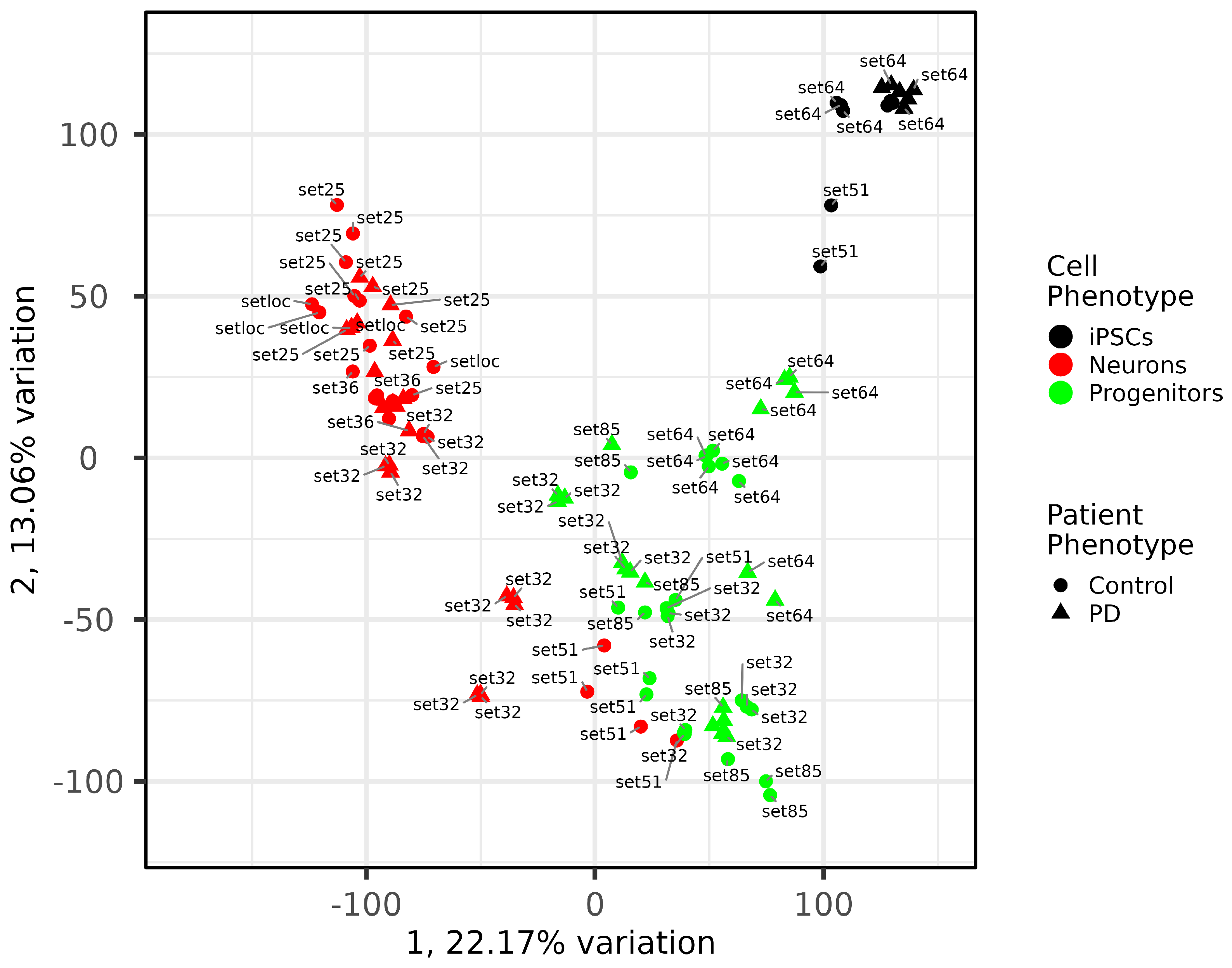
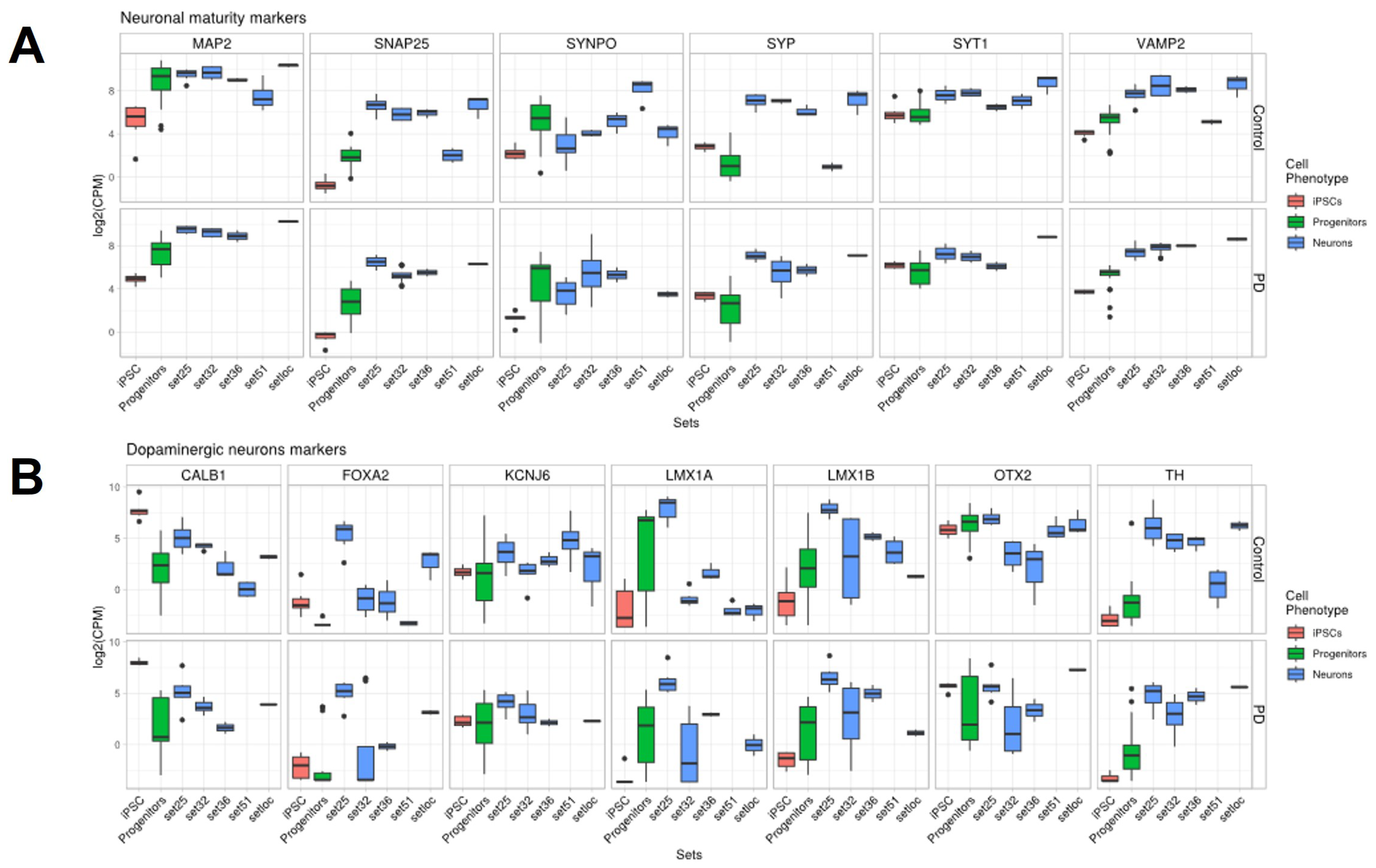
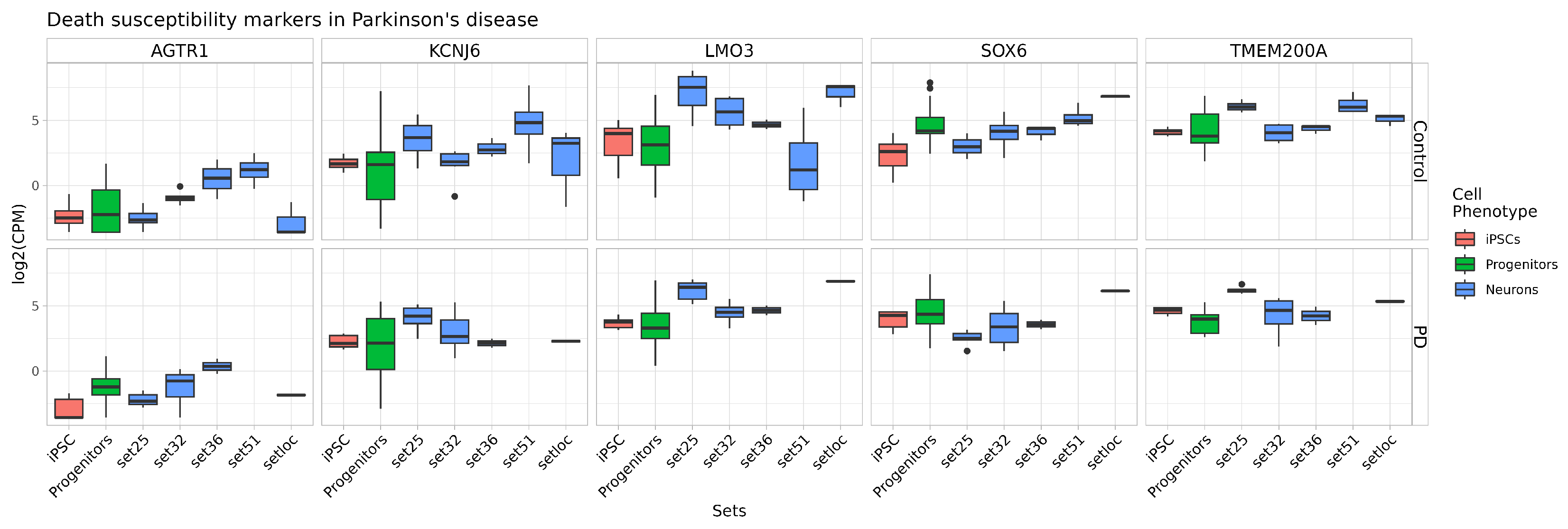
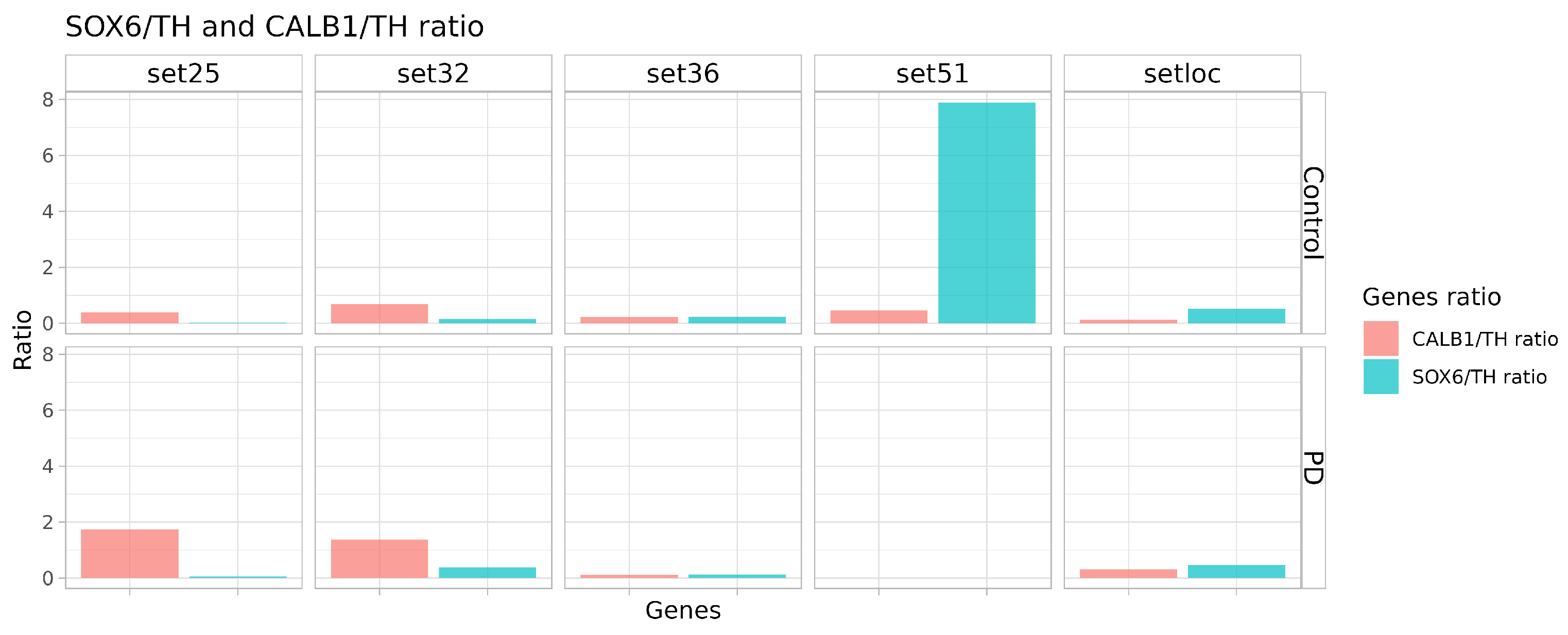
2.4. Quality and Maturity of Neurons as Confirmed by RNA Sequencing Results
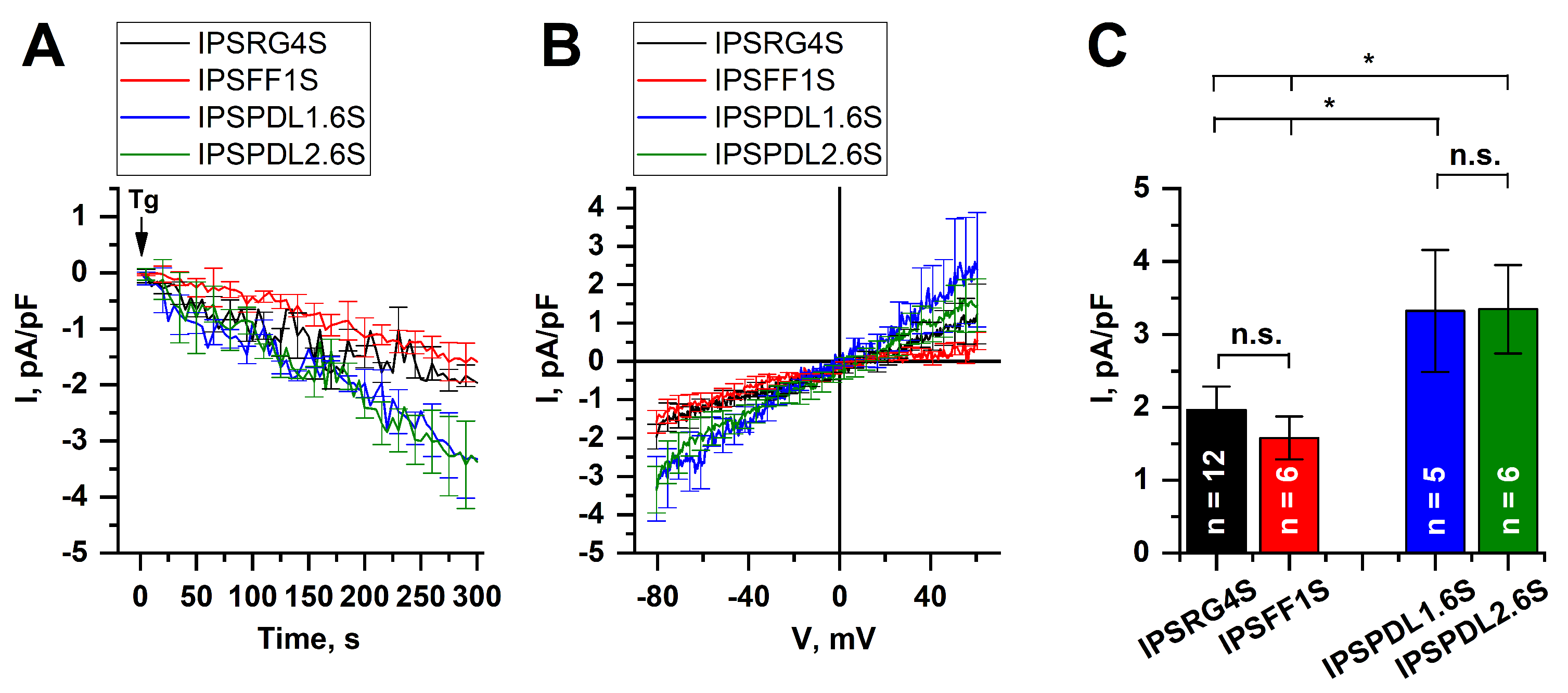
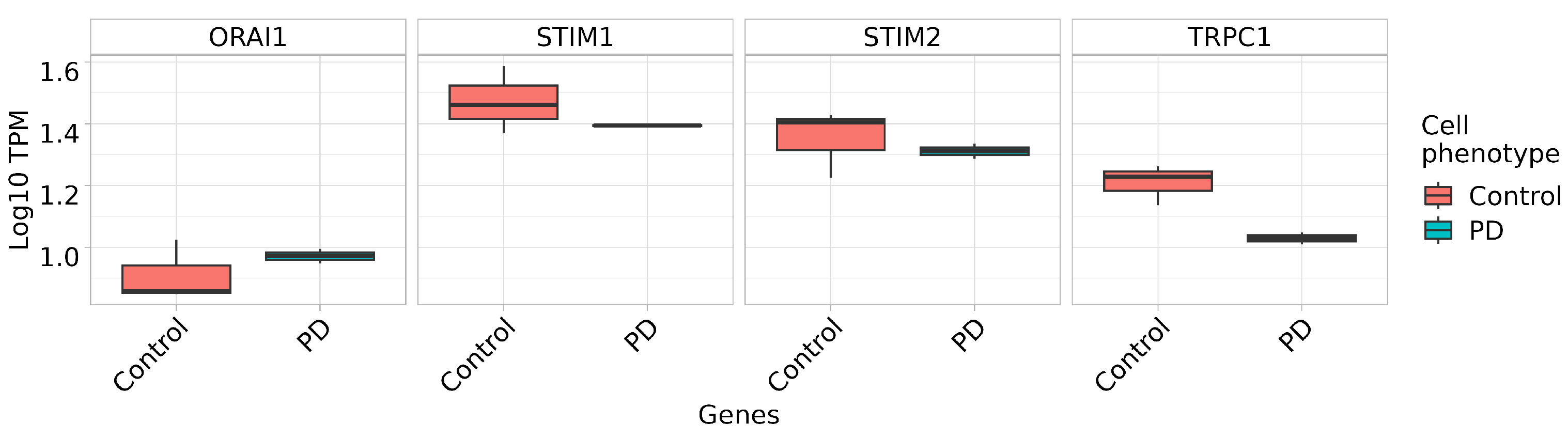
2.5. Store-Operated Calcium Entry Is Enhanced in DAns with a Mutation in the PARK8 Gene
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Recombinant Proteins and Small Molecule Compounds
4.3. Other Reagents for Cell Cultures
4.4. Molecular Biology Reagents
4.5. Consumables
4.6. Cell Culture Media Compositions
4.7. The Experiment Protocols
4.7.1. IPSCs Cultivation
4.7.2. IPSCs Differentiation to Dopaminergic Neurons
- IPSCs were detached with trypsin and plated at a density of 40,000 cells/cm in mTeSR1 or GibriS-8 medium with presence of 5 M of ROCK inhibitor.
- At a density of about 90-100%, the mTeSR1 (GibriS-8) medium was replaced by the neural differentiation medium. Cells were cultured for 14 days with the medium change every other day.
- The resulting neural progenitors were detached with Versene solution by incubating the cells for 10 min in a CO incubator at 37 C. After that, cells were centrifuged for 5 min at 250× g. The cells were washed twice with DMEM/F12 medium, each time centrifuged under the same conditions. Cells were plated at a density of 250,000–400,000 cells/cm on cell culture-treated dishes or multi-well plates (depending on the upcoming task) coated with Matrigel and cultured for 10 days in neuronal progenitor medium. The medium was changed every other day.
- The resulting ventral midbrain neuronal progenitors were detached with Versene solution as described above. Cells were plated at a density of 250,000–400,000 cells/cm on cell culture-treated dishes or multi-well plates (depending on the upcoming task) coated with Matrigel and cultured for 7 days in NB maturation medium. The medium was changed every other day. After this stage, the cells could no longer be reseeded.
- The cells were cultured in NBA maturation medium for at least 1 week until full maturity. For longer culturing, the cells continued to be maintained in the NBA maturation medium with the medium change every other day.
4.7.3. Polymerase Chain Reaction
4.7.4. Immunocytochemical Analysis
4.7.5. Flow Cytometry
Surface Marker Staining
- Mature neurons were carefully detached with Versene solution. Cells were incubated for 10 min in a CO incubator and centrifuged for 5 min at 300× g. After that, cells were washed twice with DMEM/F12 medium, centrifuged under the same conditions each time.
- A total of 500,000 cells were resuspended in 100 L PBS without calcium and magnesium supplemented with 1% FBS. Next, the primary phycoerythrin-labeled anti-N-CAM antibodies (DAKO), the primary FITC-labeled anti-CD24 antibodies (DAKO) and the corresponding isotype control antibodies (DAKO) were added in dilutions recommended by the manufacturer.
- Cells with antibodies were incubated for 1 h on ice.
- Cells were centrifuged at 300× g for 5 min at +4 °C; the supernatant was removed. Cells were washed once with PBS without calcium and magnesium supplemented with 1% FBS and centrifuged under the same conditions. The cell pellet was resuspended in neuron maturation medium (In total, 100 L of medium contained 200,000 cells). The cell suspension was mixed with an equal volume of 400 ng/mL DAPI solution and incubated for 5 minutes using ice.
- Cells were analyzed using an Acea NovoCyte 3000 flow cytometer. A total of 500,000 cells were taken per measurement.
- The obtained data were analyzed using NovoExpress software. DAPI-negative cells were isolated as a population of living cells; within this population, the proportion of N-CAM-positive and CD24-positive cells was measured.
Staining of Cytoplasmic Markers and Staining for Cell Cycle Analysis
- Mature neurons were carefully detached with Versene solution. Cells were incubated for 10 min in a CO incubator and centrifuged for 5 min at 300× g. After that, cells were washed twice with DMEM/F12 medium, centrifuged under the same conditions each time.
- Cells were resuspended using 1% PFA and incubated on ice for 15 min. Cells were washed twice with PBS without calcium and magnesium supplemented with 1% FBS. After the last wash, 100 L of washing solution was left and the cell pellet was resuspended in this volume.
- Next, 80% ethanol was carefully added to the cell suspension. After that, cells were incubated on ice for 30 min.
- The cells were washed twice with PBS without calcium and magnesium supplemented with 1% FBS.
- Each sample was resuspended in 200 L of PBS without calcium and magnesium supplemented with 1% FBS and divided into two tubes. Antibodies against TH (ab112, Abcam) were added to one part, isotype control (026102, Invitrogen) to the other. Samples were incubated at +4 °C overnight.
- Cells were washed twice using PBS without calcium and magnesium supplemented with 1% FBS. Secondary antibodies conjugated with Alexa 488 (Invitrogen) were added to the cells and incubated on ice at 30 min.
- The cells were washed twice with PBS without calcium and magnesium supplemented with 1% FBS. The cells were resuspended in the same buffer; in total, 100 L of solution contained 200,000 cells. A total of 500,000 cells were taken per measurement; an equal volume of 400 ng/mL DAPI solution was added.
- Cells were analyzed using an Acea NovoCyte 3000 flow cytometer.
- The rest cells were centrifuged and resuspended in proportion of 200,000 cells/100 L in 1 mg/mL DAPI solution and PBS with 0.1% Triton-X100. After that, cells were incubated for 30 min on ice. Next, the stained cells were used for cell cycle analysis.
- Cell cycle analysis was performed by an Acea NovoCyte 3000 flow cytometer using the Automated Cell Cycle Analysis option of NovoExpress Software. The Watson model was used for cell cycle fitting.
- The obtained data were analyzed using the NovoExpress software.
4.7.6. Measuring Dopamine Production by Neurons
4.7.7. RNA Extraction and Transcriptome Library Construction
4.7.8. Datasets Preparation
4.7.9. Fluorescent Calcium Imaging
4.7.10. Electrophysiological Recordings
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK 4,5,7 | Anaplastic lymphoma kinase 4,5,7 |
| ATP | Adenosine triphosphate |
| BDNF | Brain-derived neurotrophic factor |
| BMP | Bone morphogenic protein |
| CCCP | Carbonyl cyanide m-chlorophenylhydrazone |
| DAPI | 4,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| FBS | Fetal bovine serum |
| FGF8 | Fibroblast growth factor 8 |
| FITC | Fluorescein isothiocyanate |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GDNF | Glial-derived neurotrophic factor |
| GTP | Guanosine triphosphate |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HPLC | High-performance liquid chromatography |
| ESC | Embryonic stem cells |
| IPSC | Induced pluripotent stem cells |
| L-DOPA | 3,4-Dihydroxy-L-phenylalanin |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAP2 | Microtubule-associated protein 2 |
| MMLV | Moloney Murine Leukemia Virus |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| N-CAM | Neural cell adhesion molecule |
| NPC | Neural progenitor cells |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced kinase 1 |
| RNA | Ribonucleic acid |
| ROC | Ras-like G-domain |
| ROCK | Rho kinase |
| TGF | Transforming growth factor |
| TH | Tyrosine hydroxylase |
| SHH | Sonic hedgehog |
| VM NPC | Ventral midbrain neuronal progenitor cells |
References
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Goldman, S.; Tanner, C.M.; Yaffe, K. Traumatic brain injury in later life increases risk for P arkinson disease. Ann. Neurol. 2015, 77, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.C.; Bordelon, Y.; Bronstein, J.; Ritz, B. Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 2012, 79, 2061–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, M.H.; Farb, D.H.; Ozer, J.; Feldman, R.G.; Durso, R. Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip. Toxicol. 2014, 7, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin-Ouellet, J.; Barker, R. Parkinson’s Disease in a Dish: What Patient Specific-Reprogrammed Somatic Cells Can Tell Us about Parkinson’s Disease, If Anything? Stem Cells Int. 2012, 2012, 926147. [Google Scholar] [CrossRef] [Green Version]
- Sudhaman, S.; Prasad, K.; Behari, M.; Muthane, U.B.; Juyal, R.C.; Thelma, B. Discovery of a frameshift mutation in podocalyxin-like (PODXL) gene, coding for a neural adhesion molecule, as causal for autosomal-recessive juvenile Parkinsonism. J. Med. Genet. 2016, 53, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Premack, D. Human and animal cognition: Continuity and discontinuity. Proc. Natl. Acad. Sci. USA 2007, 104, 13861–13867. [Google Scholar] [CrossRef] [Green Version]
- Tsilidis, K.K.; Panagiotou, O.A.; Sena, E.S.; Aretouli, E.; Evangelou, E.; Howells, D.W.; Salman, R.A.S.; Macleod, M.R.; Ioannidis, J.P. Evaluation of excess significance bias in animal studies of neurological diseases. PLoS Biol. 2013, 11, e1001609. [Google Scholar] [CrossRef] [Green Version]
- Shafa, M.; Yang, F.; Fellner, T.; Rao, M.S.; Baghbaderani, B.A. Human-induced pluripotent stem cells manufactured using a current good manufacturing practice-compliant process differentiate into clinically relevant cells from three germ layers. Front. Med. 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Ásgrímsdóttir, E.S.; Arenas, E. Midbrain dopaminergic neuron development at the single cell level: In vivo and in stem cells. Front. Cell Dev. Biol. 2020, 8, 463. [Google Scholar] [CrossRef]
- Kamath, T.; Abdulraouf, A.; Burris, S.; Langlieb, J.; Gazestani, V.; Nadaf, N.M.; Balderrama, K.; Vanderburg, C.; Macosko, E.Z. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat. Neurosci. 2022, 25, 588–595. [Google Scholar] [CrossRef]
- Lagarkova, M.A.; Shutova, M.V.; Bogomazova, A.N.; Vassina, E.M.; Glazov, E.A.; Zhang, P.; Rizvanov, A.A.; Chestkov, I.V.; Kiselev, S.L. Induction of pluripotency in human endothelial cells resets epigenetic profile on genome scale. Cell Cycle 2010, 9, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, S.; Lehtonen, Š.; Chumarina, M.; Puttonen, K.A.; Azevedo, C.; Lebedeva, O.; Ruponen, M.; Oksanen, M.; Djelloul, M.; Collin, A.; et al. Creation of a library of induced pluripotent stem cells from Parkinsonian patients. npj Park. Dis. 2016, 2, 16009. [Google Scholar] [CrossRef] [Green Version]
- Bogomiakova, M.; Sekretova, E.; Eremeev, A.; Shuvalova, L.; Bobrovsky, P.; Zerkalenkova, E.; Lebedeva, O.; Lagarkova, M. Derivation of induced pluripotent stem cells line (RCPCMi007-A-1) with inactivation of the beta-2-microglobulin gene by CRISPR/Cas9 genome editing. Stem Cell Res. 2021, 55, 102451. [Google Scholar] [CrossRef]
- Nenasheva, V.; Novosadova, E.; Makarova, I.; Lebedeva, O.; Grefenshtein, M.; Arsenyeva, E.; Antonov, S.; Grivennikov, I.; Tarantul, V. The transcriptional changes of trim genes associated with Parkinson’s disease on a model of human induced pluripotent stem cells. Mol. Neurobiol. 2017, 54, 7204–7211. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.C.; Harland, R.M. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell 1992, 70, 829–840. [Google Scholar] [CrossRef]
- Smith, J.R.; Vallier, L.; Lupo, G.; Alexander, M.; Harris, W.A.; Pedersen, R.A. Inhibition of Activin/Nodal signaling promotes specification of human embryonic stem cells into neuroectoderm. Dev. Biol. 2008, 313, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Morizane, A.; Doi, D.; Kikuchi, T.; Nishimura, K.; Takahashi, J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J. Neurosci. Res. 2011, 89, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Khankhel, A.H.; Megale, H.C.; Glasauer, S.M.; Wyle, Y.; Britton, G.; Warmflash, A.; Kosik, K.S.; Siggia, E.D.; Shraiman, B.I.; et al. Human neural tube morphogenesis in vitro by geometric constraints. Nature 2021, 599, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Garrido, J.; Winn, D.; Maffezzini, C.; Wedell, A.; Freyer, C.; Falk, A.; Wredenberg, A. Protocol for the derivation, culturing, and differentiation of human iPS-cell-derived neuroepithelial stem cells to study neural differentiation in vitro. STAR Protoc. 2021, 2, 100528. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.V.; Faiz, M.; Pivonkova, H.; Khelifi, G.; Yang, H.; Gao, S.; Linderoth, E.; Zhen, M.; Karadottir, R.T.; Hussein, S.M.; et al. Signal requirement for cortical potential of transplantable human neuroepithelial stem cells. Nat. Commun. 2022, 13, 2844. [Google Scholar] [CrossRef]
- Gilbert, S.F. Developmental Biology; Sinauer Associates, Inc.: Sunderland, MA, USA, 2003. [Google Scholar]
- Wurst, W.; Bally-Cuif, L. Neural plate patterning: Upstream and downstream of the isthmic organizer. Nat. Rev. Neurosci. 2001, 2, 99–108. [Google Scholar] [CrossRef]
- Li, X.J.; Hu, B.Y.; Jones, S.A.; Zhang, Y.S.; LaVaute, T.; Du, Z.W.; Zhang, S.C. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells 2008, 26, 886–893. [Google Scholar] [CrossRef] [Green Version]
- Hochbaum, D.; Barila, G.; Ribeiro-Neto, F.; Altschuler, D.L. Radixin assembles cAMP effectors Epac and PKA into a functional cAMP compartment: Role in cAMP-dependent cell proliferation. J. Biol. Chem. 2011, 286, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Wiedenmann, B.; Franke, W.W. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 1985, 41, 1017–1028. [Google Scholar] [CrossRef]
- Edelmann, L.; Hanson, P.; Chapman, E.; Jahn, R. Synaptobrevin binding to synaptophysin: A potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995, 14, 224–231. [Google Scholar] [CrossRef]
- Sarnat, H.B. Clinical neuropathology practice guide 5-2013: Markers of neuronal maturation. Clin. Neuropathol. 2013, 32, 340. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Goridis, C.; Brunet, J.F. NCAM: Structural diversity, function and regulation of expression. In Seminars in Cell Biology; Elsevier: Amsterdam, The Netherlands, 1992; Volume 3, pp. 189–197. [Google Scholar]
- Yuan, S.H.; Martin, J.; Elia, J.; Flippin, J.; Paramban, R.I.; Hefferan, M.P.; Vidal, J.G.; Mu, Y.; Killian, R.L.; Israel, M.A.; et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS ONE 2011, 6, e17540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuruş, M.; Akbari, S.; Eskier, D.; Bursalı, A.; Ergin, K.; Erdal, E.; Karakülah, G. Transcriptome dynamics of human neuronal differentiation from iPSC. Front. Cell Dev. Biol. 2021, 9, 3477. [Google Scholar] [CrossRef]
- Novosadova, E.; Anufrieva, K.; Kazantseva, E.; Arsenyeva, E.; Fedoseyeva, V.; Stepanenko, E.; Poberezhniy, D.; Illarioshkin, S.; Novosadova, L.; Gerasimova, T.; et al. Transcriptome datasets of neural progenitors and neurons differentiated from induced pluripotent stem cells of healthy donors and Parkinson’s disease patients with mutations in the PARK2 gene. Data Brief 2022, 41, 107958. [Google Scholar] [CrossRef]
- Vlasov, I.N.; Alieva, A.K.; Novosadova, E.V.; Arsenyeva, E.L.; Rosinskaya, A.V.; Partevian, S.A.; Grivennikov, I.A.; Shadrina, M.I. Transcriptome Analysis of Induced Pluripotent Stem Cells and Neuronal Progenitor Cells, Derived from Discordant Monozygotic Twins with Parkinson’s Disease. Cells 2021, 10, 3478. [Google Scholar] [CrossRef]
- Woodard, C.M.; Campos, B.A.; Kuo, S.-H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.; Paull, D.; et al. iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, H.J.; Patikas, N.; Foskolou, S.; Field, S.F.; Park, J.E.; Byrne, M.L.; Bassett, A.R.; Metzakopian, E. Single-cell transcriptomics of Parkinson’s disease human in vitro models reveals dopamine neuron-specific stress responses. Cell Rep. 2020, 33, 108263. [Google Scholar] [CrossRef]
- Fiorenzano, A.; Sozzi, E.; Parmar, M.; Storm, P. Dopamine neuron diversity: Recent advances and current challenges in human stem cell models and single cell sequencing. Cells 2021, 10, 1366. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Grekhnev, D.A.; Novikova, I.V.; Krisanova, A.V.; Yuskovets, V.N.; Chernov, N.M.; Yakovlev, I.P.; Kaznacheyeva, E.V.; Vigont, V.A. Dithiadiazole derivative 3-(4-nitrophenyl)-5-phenyl-3H-1,2,3,4-dithiadiazole-2-oxide - Novel modulator of store-operated calcium entry. Biochem. Biophys. Res. Commun. 2022, 626, 38–43. [Google Scholar] [CrossRef]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Czeredys, M.; Vigont, V.A.; Boeva, V.A.; Mikoshiba, K.; Kaznacheyeva, E.V.; Kuznicki, J. Huntingtin-associated protein 1A regulates store-operated calcium entry in medium spiny neurons from transgenic YAC128 mice, a model of Huntington’s disease. Front. Cell. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef]
- Latoszek, E.; Czeredys, M. Molecular Components of Store-Operated Calcium Channels in the Regulation of Neural Stem Cell Physiology, Neurogenesis, and the Pathology of Huntington’s Disease. Front. Cell Dev. Biol. 2021, 9, 657337. [Google Scholar] [CrossRef]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E. Both Orai1 and TRPC1 are involved in excessive store-operated calcium entry in striatal neurons expressing mutant huntingtin exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef] [Green Version]
- Vigont, V.; Nekrasov, E.; Shalygin, A.; Gusev, K.; Klushnikov, S.; Illarioshkin, S.; Lagarkova, M.; Kiselev, S.L.; Kaznacheyeva, E. Patient-specific iPSC-based models of Huntington’s disease as a tool to study store-operated calcium entry drug targeting. Front. Pharmacol. 2018, 9, 696. [Google Scholar] [CrossRef]
- Vigont, V.A.; Grekhnev, D.A.; Lebedeva, O.S.; Gusev, K.O.; Volovikov, E.A.; Skopin, A.Y.; Bogomazova, A.N.; Shuvalova, L.D.; Zubkova, O.A.; Khomyakova, E.A.; et al. STIM2 mediates excessive store-operated calcium entry in patient-specific iPSC-derived neurons modeling a juvenile form of Huntington’s disease. Front. Cell Dev. Biol. 2021, 9, 625231. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greggio, E.; Bisaglia, M.; Civiero, L.; Bubacco, L. Leucine-rich repeat kinase 2 and alpha-synuclein: Intersecting pathways in the pathogenesis of Parkinson’s disease? Mol. Neurodegener. 2011, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Cookson, M.R. Role of LRRK2 kinase dysfunction in Parkinson disease. Expert Rev. Mol. Med. 2011, 13, e20. [Google Scholar] [CrossRef] [PubMed]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial Dysfunction in Parkinson’s Disease. Park. Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Familial Parkinson’s disease iPSCs show cellular deficits in mitochondrial responses that can be pharmacologically rescued. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [Green Version]
- Cobb, M.M.; Ravisankar, A.; Skibinski, G.; Finkbeiner, S. iPS cells in the study of PD molecular pathogenesis. Cell Tissue Res. 2018, 373, 61–77. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.C.; Qi, X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef] [Green Version]
- Korecka, J.A.; Thomas, R.; Christensen, D.P.; Hinrich, A.J.; Ferrari, E.J.; Levy, S.A.; Hastings, M.L.; Hallett, P.J.; Isacson, O. Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson’s disease patient fibroblasts. Hum. Mol. Genet. 2019, 28, 3232–3243. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.; Martin, I.; Xiong, Y.; Eacker, S.M.; Ingolia, N.T.; Dawson, T.M.; Dawson, V.L. Dysregulated mRNA translation in the G2019S LRRK2 and LRRK2 knock-out mouse brains. Eneuro 2021, 8, ENEURO.0310-21.2021. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.; et al. Defects in mRNA translation in LRRK2-mutant hiPSC-derived dopaminergic neurons lead to dysregulated calcium homeostasis. Cell Stem Cell 2020, 27, 633–645. [Google Scholar] [CrossRef]
- Zampese, E.; Wokosin, D.L.; Gonzalez-Rodriguez, P.; Guzman, J.N.; Tkatch, T.; Kondapalli, J.; Surmeier, W.C.; D’Alessandro, K.B.; De Stefani, D.; Rizzuto, R.; et al. Ca2+ channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci. Adv. 2022, 8, eabp8701. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Spät, A.; Szanda, G. The role of mitochondria in the activation/maintenance of SOCE: Store-operated Ca 2+ entry and mitochondria. In Store-Operated Ca2+ Entry (SOCE) Pathways: Emerging Signaling Concepts in Human (Patho) Physiology; Springer: Berlin, Germany, 2017; pp. 257–275. [Google Scholar]
- Villalobos, C.; Gutiérrez, L.G.; Hernández-Morales, M.; Del Bosque, D.; Núñez, L. Mitochondrial control of store-operated Ca2+ channels in cancer: Pharmacological implications. Pharmacol. Res. 2018, 135, 136–143. [Google Scholar] [CrossRef]
- Nan, J.; Li, J.; Lin, Y.; Saif Ur Rahman, M.; Li, Z.; Zhu, L. The interplay between mitochondria and store-operated Ca2+ entry: Emerging insights into cardiac diseases. J. Cell. Mol. Med. 2021, 25, 9496–9512. [Google Scholar] [CrossRef]
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Jiang, Q.; Zhao, J.; Ren, Y.; Sutton, M.D.; Feng, J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J. Biol. Chem. 2005, 280, 17154–17162. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Hubschman, J.P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Consiglio, A.; Richaud, Y.; Rodríguez-Pizà, I.; Dehay, B.; Edel, M.; Bove, J.; Memo, M.; Vila, M.; Raya, A.; et al. Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of LMX1A in human embryonic stem cells and induced pluripotent stem cells. Hum. Gene Ther. 2012, 23, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Nefzger, C.M.; Su, C.T.; Fabb, S.A.; Hartley, B.J.; Beh, S.J.; Zeng, W.R.; Haynes, J.M.; Pouton, C.W. Lmx1a allows context-specific isolation of progenitors of GABAergic or dopaminergic neurons during neural differentiation of embryonic stem cells. Stem Cells 2012, 30, 1349–1361. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Bading, H.; Ginty, D.D.; Greenberg, M.E. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 1993, 260, 181–186. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K. PCAtools: PCAtools: Everything Principal Components Analysis. R Package Version 1.1. 10. 2019. Available online: http://www.bioconductor.org/packages/devel/bioc/vignettes/PCAtools/inst/doc/PCAtools.html (accessed on 30 March 2023).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 30 March 2023). [CrossRef]













| Cell Line | pg of Dopamine /10 TH+ Cells (Mean ± SEM) |
|---|---|
| IPSRG2L (Healthy) | 6.99 |
| IPSRG4S (Healthy) | 73.4 |
| IPSFF1S (Healthy) | 25.55±1.22 (n = 2) |
| IPSPDL1.6S (mutation in PARK8 gene) | 51.49 ± 52.28 (n = 3) |
| IPSPDL2.15L (mutation in PARK8 gene) | 61.21 ± 18.16 (n = 2) |
| IPSPDL2.6S (mutation in PARK8 gene) | 142.45 ± 63.85 (n = 2) |
| IPSPDP1.5L (mutation in PARK2 gene) | 72.06 ± 82.04 (n = 2) |
| Author | BioProject | Instrument | PE/SE | Length | Library Type | N | iPSCs | Progenitors | Neurons | Alias |
|---|---|---|---|---|---|---|---|---|---|---|
| [35] | PRJNA330836 | HiSeq 2000 | PAIRED | 50 | polyA | 5 | 0|0 | 0|0 | 3|2 | set36 |
| [36] | PRJNA699151 | NextSeq 500 | PAIRED | 40 | unk | 10 | 2|0 | 4|0 | 4|0 | set51 |
| PRJNA761085 | NextSeq 500 | PAIRED | 100 | rRNAdepl | 10 | 0|0 | 5|5 | 0|0 | set85 | |
| [37] | PRJNA750432 | NextSeq 500 | PAIRED | 75 | polyA | 36 | 0|0 | 9|9 | 6|12 | set32 |
| [38] | PRJNA767364 | HiSeq 1500 | SINGLE | 50 | polyA | 24 | 6|6 | 6|6 | 0|0 | set64 |
| [39] | PRJNA264625 | HiSeq 2500 | SINGLE | 100 | polyA | 14 | 0|0 | 0|0 | 8|6 | set25 |
| Our data | NA | Novaseq 6000 | PAIRED | 100 | polyA | 5 | 0|0 | 0|0 | 3|2 | setloc |
| Gene | Primer Sequence | T |
|---|---|---|
| TH | 5 CCAAGCAGGCAGAGGCCATCATGT 3 | 60 °C, 30 cycles |
| 5 GGCGTAGAGGCCCTTCAGCGT 3 | ||
| TH (for qPCR) | 5 GGGCTGTGTAAGCAGAACG 3 | 60 °C, 45 cycles |
| 5 AAGGCCCGAATCTCAGGCT 3 | ||
| SYP | 5 GCTTTGTGAAGGTGCTGCAA 3 | 60 °C, 30 cycles |
| 5 GCCTGAAGGGGTACTCGAAC 3 | ||
| GAPDH | 5 GAAGGTGAAGGTCGGAGTCA 3 | 60 °C, 25 cycles |
| 5 TTCACACCCATGACGAACAT 3 | ||
| GAPDH (for qPCR) | 5 5’ GAAGGTGAAGGTCGGAGTCA 3 | 60 °C, 45 cycles |
| 5 GTTGAGGTCAATGAAGGGGTC 3 | ||
| Oct4 (for qPCR) | 5 CAAAGCAGAAACCCTCGTGC 3 | 60 °C, 45 cycles |
| 5 TGATCTGCTGCAGTGTGGG 3 | ||
| Tubb3 (for qPCR) | 5 GGCCAAGGGTCACTACACG 3 | 60 °C, 45 cycles |
| 5 GCAGTCGCAGTTTTCACACTC 3 | ||
| DAT1 (for qPCR) | 5 TGTGGGCTTCACGGTCATC 3 | 60 °C, 45 cycles |
| 5 GTCCCAAAAGTGTCGTTGAGG 3 |
| Antigen | Dilution | Cat# and Manufacturer |
|---|---|---|
| Oct4 | 1:200 | ab18976, Abcam |
| Sox1 | 1:500 | ab87775, Abcam |
| Pax6 | 1:50 | ab78545, Abcam |
| -III-tubulin | 1:1000 | ab18207, Abcam |
| TH | 1:2000 | ab112, Abcam |
| Goat-anti-Rabbit IgG | 1:800 | ab181474, Abcam |
| Goat-anti-Rabbit IgG | 1:800 | A32732, Thermo Fisher |
| Goat anti-Mouse IgG (H+L) | 1:800 | A11001, Thermo Fisher |
| Goat anti-Mouse IgG (H+L) | 1:800 | A21422, Thermo Fisher |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebedeva, O.S.; Sharova, E.I.; Grekhnev, D.A.; Skorodumova, L.O.; Kopylova, I.V.; Vassina, E.M.; Oshkolova, A.; Novikova, I.V.; Krisanova, A.V.; Olekhnovich, E.I.; et al. An Efficient 2D Protocol for Differentiation of iPSCs into Mature Postmitotic Dopaminergic Neurons: Application for Modeling Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 7297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087297
Lebedeva OS, Sharova EI, Grekhnev DA, Skorodumova LO, Kopylova IV, Vassina EM, Oshkolova A, Novikova IV, Krisanova AV, Olekhnovich EI, et al. An Efficient 2D Protocol for Differentiation of iPSCs into Mature Postmitotic Dopaminergic Neurons: Application for Modeling Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(8):7297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087297
Chicago/Turabian StyleLebedeva, Olga S., Elena I. Sharova, Dmitriy A. Grekhnev, Liubov O. Skorodumova, Irina V. Kopylova, Ekaterina M. Vassina, Arina Oshkolova, Iuliia V. Novikova, Alena V. Krisanova, Evgenii I. Olekhnovich, and et al. 2023. "An Efficient 2D Protocol for Differentiation of iPSCs into Mature Postmitotic Dopaminergic Neurons: Application for Modeling Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 8: 7297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087297