
Inhibition of Bromodomain Proteins Enhances Oncolytic HAdVC5 Replication and Efficacy in Pancreatic Ductal Adenocarcinoma (PDAC) Models
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Optimization of Conditions for High-Throughput Screening (HTS)
2.2. High Throughput Screening (HTS) of a Small Molecule Compound Library
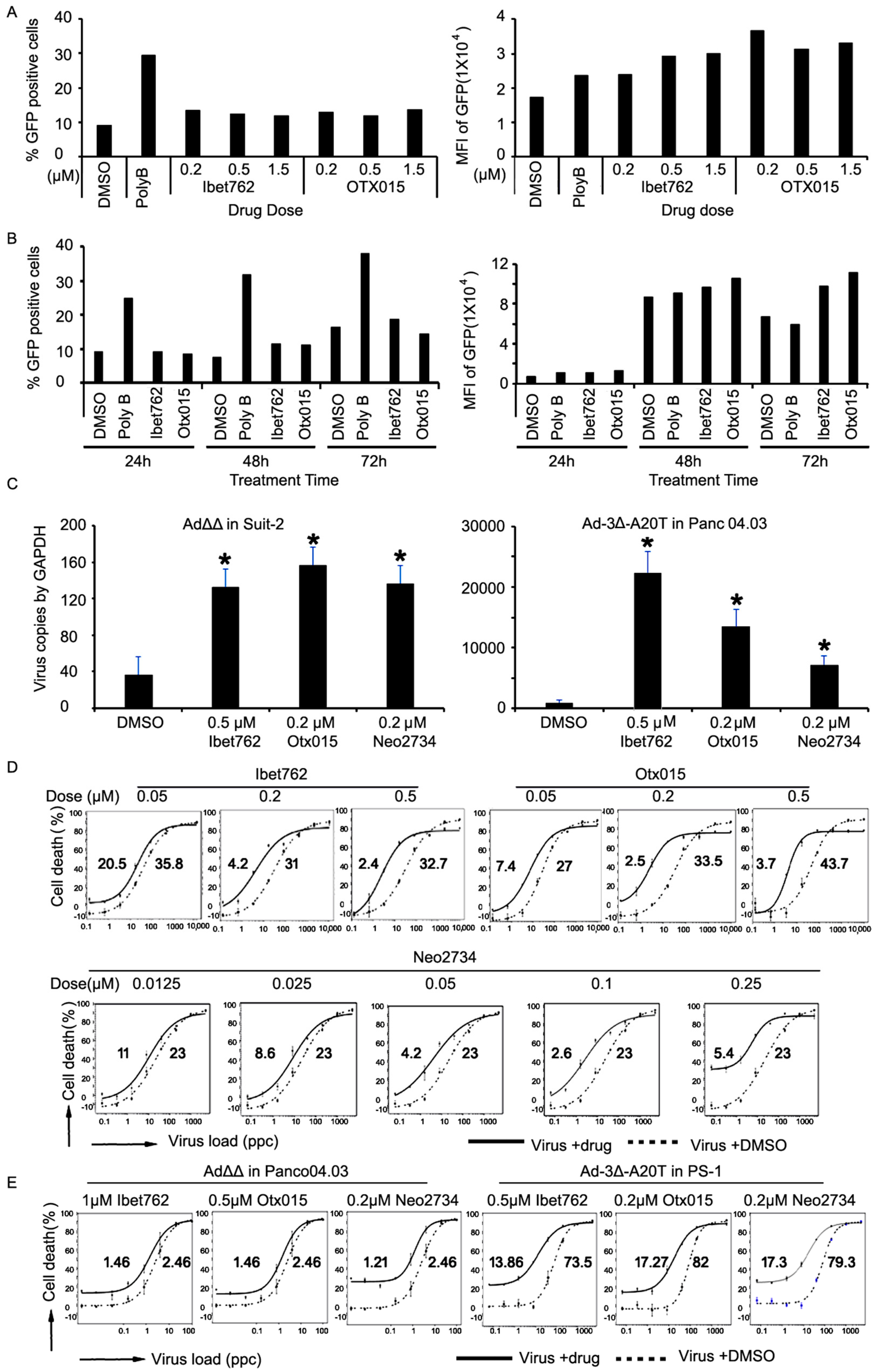
2.3. The Bromodomain Inhibitors OTX-015 and iBet-762 Promote Virus Replication Rather Than Uptake
2.4. Virus-Induced Cancer Cell Killing Is Enhanced by the OTX-015, iBet-762, and Neo-2734 Inhibitors
2.5. Expression of c-Myc Is Greatly Reduced in Cells Treated with the OTX-015, iBet-762, or Neo-2734 Inhibitors
2.6. The Ad∆∆ Mutant in Combination with Neo-2734 Improved on In Vivo Efficacy in Suit-2 Xenograft Models
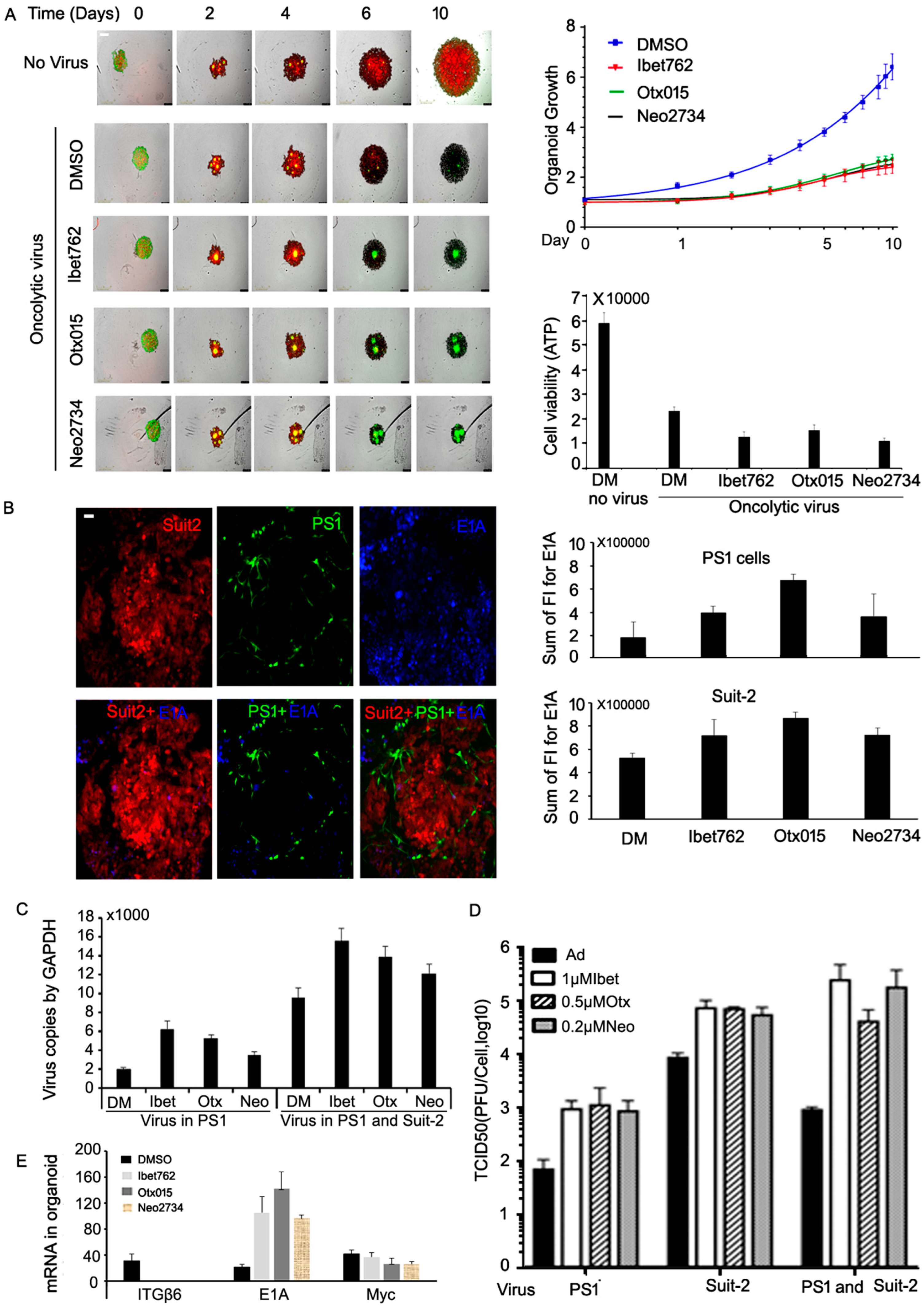
2.7. All Three Bromodomain Inhibitors Promote Virus Spread and Cell Killing in 3D Co-Cultures of Suit-2 Cells and PS-1 Cancer-Associated Stromal Cells
2.8. Neo-2734 Enhanced Adenoviral Gene Expression and Replication In Vivo
2.9. OTX-015, iBet-762, and Neo-2734 Inhibitors Affect Both Viral Gene Expression and Regulation of Cellular Growth and Immune Factors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Adenoviruses
4.2. High Throughput Screening (HTS) Assay for Adenoviral Replication
4.3. DNA Extraction and qPCR Validation
4.4. Viral Replication Assay by Tissue Culture Infections Dose (TCID50)
4.5. Cell Viability Assay
4.6. Immunoblotting of Nuclear and Cytosolic Proteins
4.7. Isolation of RNA and Analysis of mRNA Expression
4.8. Organoid Co-Cultures
4.9. In Vivo Tumour Growth and Bio-Distribution
4.10. RNA Sequencing
4.11. RNA-Seq Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H. Progress in the knowledge and treatment of advanced pancreatic cancer: From benchside to bedside. Cancer Treat. Rev. 2014, 40, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the management of pancreatic cancer: Surgery is not enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Radon, T.P.; Massat, N.J.; Jones, R.; Alrawashdeh, W.; Dumartin, L.; Ennis, D.; Duffy, S.W.; Kocher, H.M.; Pereira, S.P.; Guarner, L.; et al. Identification of a Three-Biomarker Panel in Urine for Early Detection of Pancreatic Adenocarcinoma. Clin. Cancer Res. 2015, 21, 3512–3521. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, S.; Massat, N.J.; Radon, T.P.; Sangaralingam, A.; Banissi, A.; Ennis, D.P.; Dowe, T.; Chelala, C.; Pereira, S.P.; Kocher, H.M.; et al. Noninvasive urinary miRNA biomarkers for early detection of pancreatic adenocarcinoma. Am. J. Cancer Res. 2015, 5, 3455–3466. [Google Scholar] [PubMed]
- Araki, H.; Tazawa, H.; Kanaya, N.; Kajiwara, Y.; Yamada, M.; Hashimoto, M.; Kikuchi, S.; Kuroda, S.; Yoshida, R.; Umeda, Y.; et al. Oncolytic virus-mediated p53 overexpression promotes immunogenic cell death and efficacy of PD-1 blockade in pancreatic cancer. Mol. Ther. Oncolytics 2022, 27, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, Z.; Li, L.; Wu, J.; Zhang, H.; Zhang, H.; Lei, T.; Xu, B. Oncolytic Adenovirus: Prospects for Cancer Immunotherapy. Front. Microbiol. 2021, 12, 707290. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Bazan-Peregrino, M.; Gil-Martin, M.; Alvarez, R.; Macarulla, T.; Riesco-Martinez, M.C.; Verdaguer, H.; Guillen-Ponce, C.; Farrera-Sal, M.; Moreno, R.; et al. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J. Immunother. Cancer 2022, 10, e003255. [Google Scholar] [CrossRef]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Stahle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H.; et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Waddington, S.N.; Buckley, S.M.; Custers, J.; Havenga, M.J.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Effect of neutralizing sera on factor x-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009, 83, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Leitner, S.; Sweeney, K.; Oberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Hallden, G. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin. Cancer Res. 2009, 15, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blazquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Hallden, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer Ther. 2018, 17, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; Tazawa, H.; Yamada, M.; Kanaya, N.; Fushimi, T.; Kikuchi, S.; Kuroda, S.; Ohara, T.; Noma, K.; Yoshida, R.; et al. Oncolytic virus-mediated reducing of myeloid-derived suppressor cells enhances the efficacy of PD-L1 blockade in gemcitabine-resistant pancreatic cancer. Cancer Immunol. Immunother. 2023, 72, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sal, M.; Moreno, R.; Mato-Berciano, A.; Maliandi, M.V.; Bazan-Peregrino, M.; Alemany, R. Hyaluronidase expression within tumors increases virotherapy efficacy and T cell accumulation. Mol. Ther. Oncolytics 2021, 22, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Oberg, D.; Yanover, E.; Adam, V.; Sweeney, K.; Costas, C.; Lemoine, N.R.; Hallden, G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin. Cancer Res. 2010, 16, 541–553. [Google Scholar] [CrossRef]
- Pantelidou, C.; Cherubini, G.; Lemoine, N.R.; Hallden, G. The E1B19K-deleted oncolytic adenovirus mutant AdDelta19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget 2016, 7, 15703–15724. [Google Scholar] [CrossRef]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef]
- Coughlan, L.; Vallath, S.; Gros, A.; Gimenez-Alejandre, M.; Van Rooijen, N.; Thomas, G.J.; Baker, A.H.; Cascallo, M.; Alemany, R.; Hart, I.R. Combined fiber modifications both to target αvβ6 and detarget the coxsackievirus-adenovirus receptor improve virus toxicity profiles in vivo but fail to improve antitumoral efficacy relative to adenovirus serotype 5. Hum. Gene Ther. 2012, 23, 960–979. [Google Scholar] [CrossRef]
- Hausner, S.H.; Abbey, C.K.; Bold, R.J.; Gagnon, M.K.; Marik, J.; Marshall, J.F.; Stanecki, C.E.; Sutcliffe, J.L. Targeted in vivo imaging of integrin alphavbeta6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009, 69, 5843–5850. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.A.; Marlow, G.; Uusi-Kerttula, H.K.; Seaton, G.; Piggott, L.; Badder, L.M.; Clarkson, R.W.E.; Chester, J.D.; Parker, A.L. Efficient Intravenous Tumor Targeting Using the alphavbeta6 Integrin-Selective Precision Virotherapy Ad5NULL-A20. Viruses 2021, 13, 864. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Pennella, M.A.; Zemke, N.R.; Eng, C.; Berk, A.J. Adenovirus E1A Activation Domain Regulates H3 Acetylation Affecting Varied Steps in Transcription at Different Viral Promoters. J. Virol. 2018, 92, 10–128. [Google Scholar] [CrossRef]
- Zemke, N.R.; Gou, D.; Berk, A.J. Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes. Dev. 2019, 33, 828–843. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Su, T.; Li, B.; Bonora, G.; Oberai, A.; Chan, Y.; Sasidharan, R.; Berk, A.J.; Pellegrini, M.; Kurdistani, S.K. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012, 22, 1212–1221. [Google Scholar] [CrossRef]
- Rodriguez, M.; Rodriguez, I.G.; Nattress, C.; Qureshi, A.; Hallden, G. HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models. Viruses 2022, 14, 1006. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Ryan, K.R.; Giles, F.; Morgan, G.J. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur. J. Haematol. 2021, 106, 90–99. [Google Scholar] [CrossRef]
- Baldan, F.; Allegri, L.; Lazarevic, M.; Catia, M.; Milosevic, M.; Damante, G.; Milasin, J. Biological and molecular effects of bromodomain and extra-terminal (BET) inhibitors JQ1, IBET-151, and IBET-762 in OSCC cells. J. Oral. Pathol. Med. 2019, 48, 214–221. [Google Scholar] [CrossRef]
- Graziani, V.; Garcia, A.R.; Alcolado, L.S.; Le Guennec, A.; Henriksson, M.A.; Conte, M.R. Metabolic rewiring in MYC-driven medulloblastoma by BET-bromodomain inhibition. Sci. Rep. 2023, 13, 1273. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Hino, S.; Usuki, S.; Miyazaki, Y.; Oda, T.; Nakao, M.; Ito, T.; Yamagata, K. YAP/BRD4-controlled ROR1 promotes tumor-initiating cells and hyperproliferation in pancreatic cancer. EMBO J. 2023, 42, e112614. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef]
- Karim, R.; Tse, G.; Putti, T.; Scolyer, R.; Lee, S. The significance of the Wnt pathway in the pathology of human cancers. Pathology 2004, 36, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin alphavbeta6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019, 249, 332–342. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Gaudio, E.; Tarantelli, C.; Ponzoni, M.; Odore, E.; Rezai, K.; Bernasconi, E.; Cascione, L.; Rinaldi, A.; Stathis, A.; Riveiro, E.; et al. Bromodomain inhibitor OTX015 (MK-8628) combined with targeted agents shows strong in vivo antitumor activity in lymphoma. Oncotarget 2016, 7, 58142–58147. [Google Scholar] [CrossRef]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-specific DNA binding by the c-Myc protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Li, J.; Li, M.; Zhuo, Y.; Ren, K.; Li, E.; Yang, G. Enhancement of adenovirus infection and adenoviral vector-mediated gene delivery by bromodomain inhibitor JQ1. Sci. Rep. 2018, 8, 11554. [Google Scholar] [CrossRef] [PubMed]
- Pelka, P.; Ablack, J.N.; Shuen, M.; Yousef, A.F.; Rasti, M.; Grand, R.J.; Turnell, A.S.; Mymryk, J.S. Identification of a second independent binding site for the pCAF acetyltransferase in adenovirus E1A. Virology 2009, 391, 90–98. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Tansey, W.P. Adenoviral E1A function through Myc. Cancer Res. 2009, 69, 6–9. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e1314. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Lun, A.T.; Chen, Y.; Smyth, G.K. It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol. Biol. 2016, 1418, 391–416. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, T.; Symonds, A.; Hickman, O.J.; Wu, D.; Wang, P.; Lemoine, N.; Wang, Y.; Linardopoulos, S.; Halldén, G. Inhibition of Bromodomain Proteins Enhances Oncolytic HAdVC5 Replication and Efficacy in Pancreatic Ductal Adenocarcinoma (PDAC) Models. Int. J. Mol. Sci. 2024, 25, 1265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021265
Miao T, Symonds A, Hickman OJ, Wu D, Wang P, Lemoine N, Wang Y, Linardopoulos S, Halldén G. Inhibition of Bromodomain Proteins Enhances Oncolytic HAdVC5 Replication and Efficacy in Pancreatic Ductal Adenocarcinoma (PDAC) Models. International Journal of Molecular Sciences. 2024; 25(2):1265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021265
Chicago/Turabian StyleMiao, Tizong, Alistair Symonds, Oliver J. Hickman, Dongsheng Wu, Ping Wang, Nick Lemoine, Yaohe Wang, Spiros Linardopoulos, and Gunnel Halldén. 2024. "Inhibition of Bromodomain Proteins Enhances Oncolytic HAdVC5 Replication and Efficacy in Pancreatic Ductal Adenocarcinoma (PDAC) Models" International Journal of Molecular Sciences 25, no. 2: 1265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021265