Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells


Abstract
:Introduction
Materials and Methods
Chemicals and Cell Culture
Cell Treatment
Cell Treatment for Proliferation
Cell Treatment for Cytotoxicity
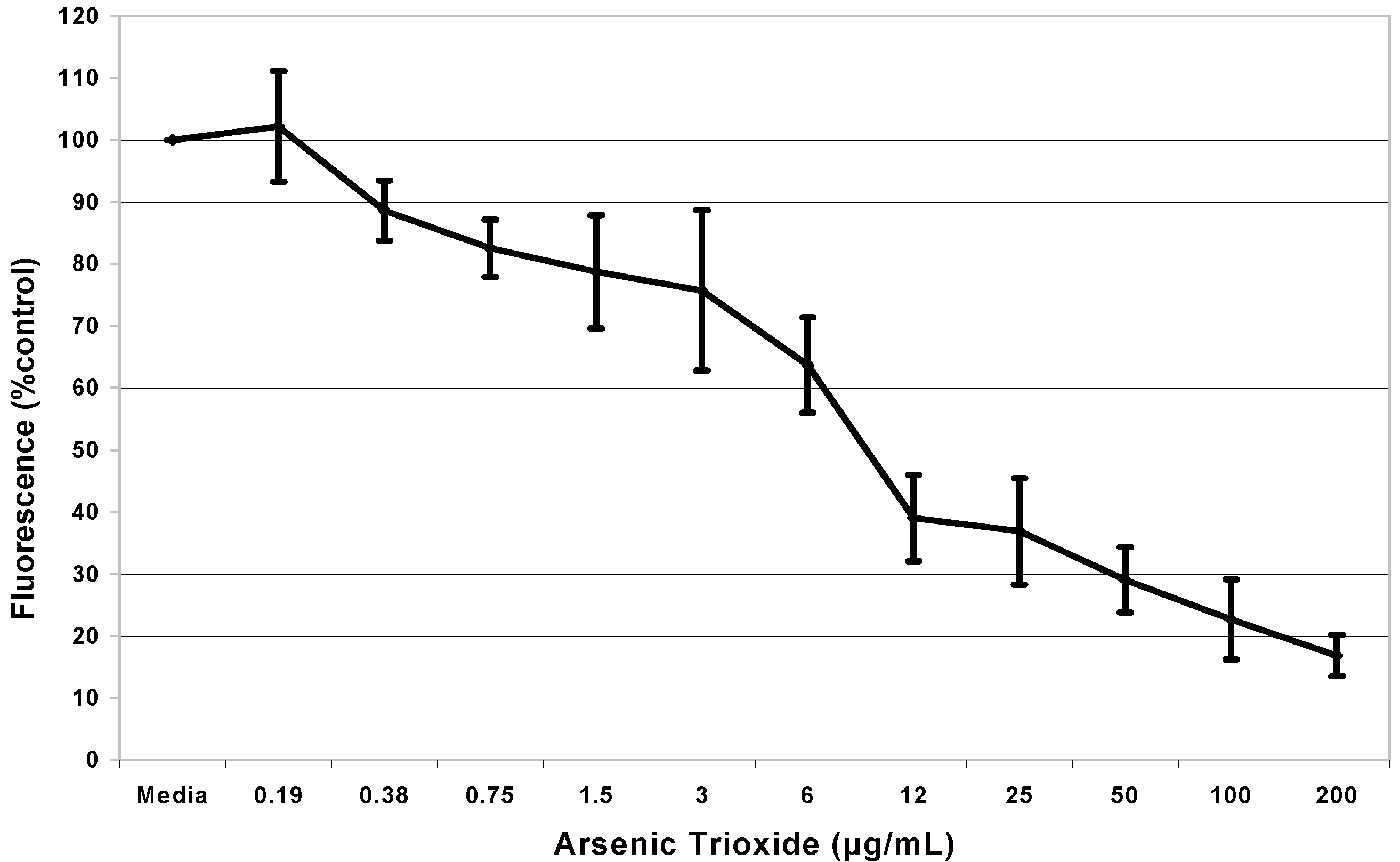
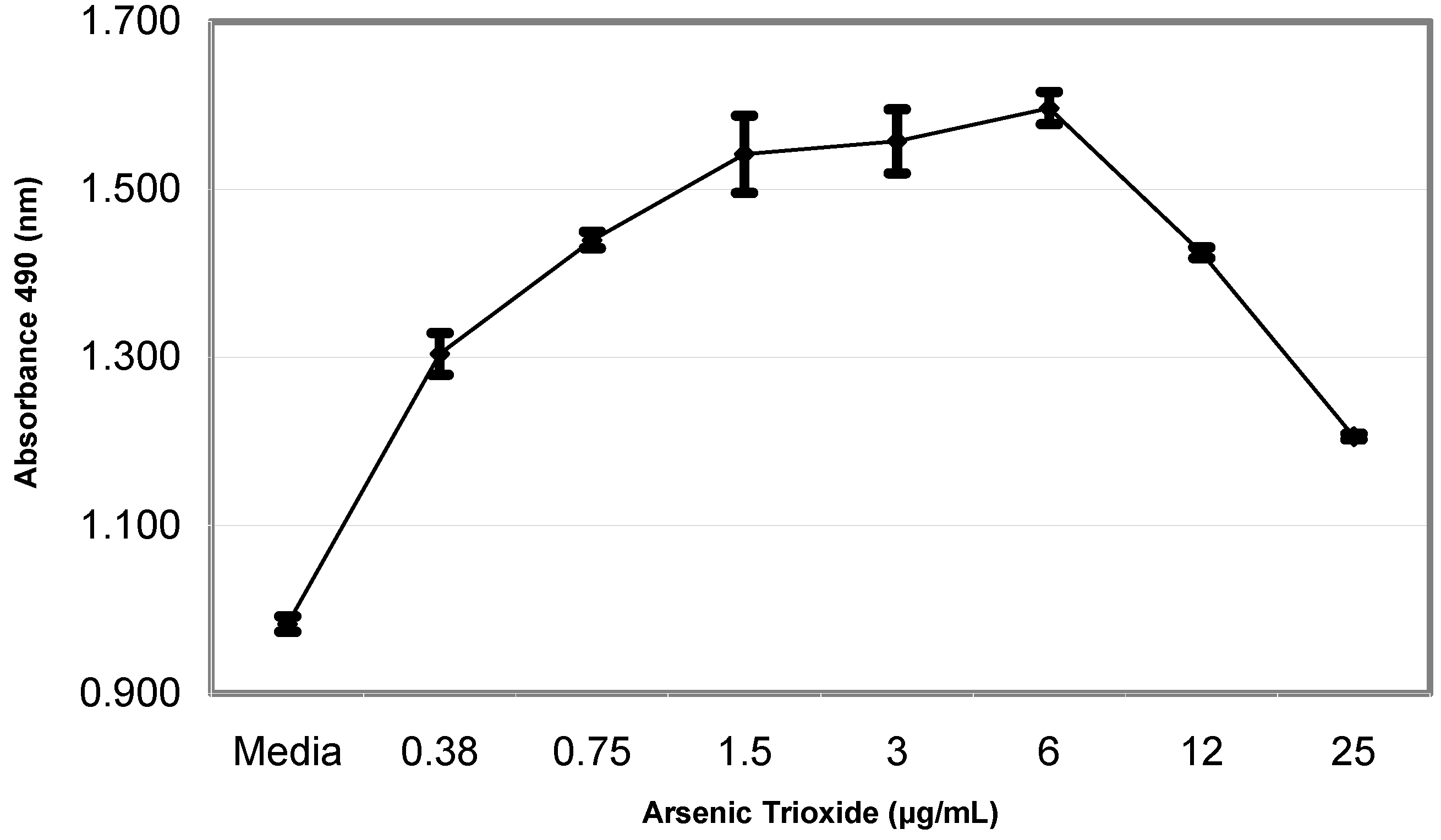
Results

{kind=link}
{kind=link}
| Cell Types | Cytotoxicity (LD50) | Peak Proliferation Dose |
| Keratinocytes (HaCaT cells) | 9 μg/mL (45.5µM) | 6 μg/mL (30µM) |
| Melanocytes (CRL 1675) | 1.5 μg/mL (7.6µM) | 0.19 μg/mL (0.95µM) |
| Dendritic cells | 1.5 μg/mL (7.6µM) | 0.19 μg/mL (0.95µM) |
| Dermal fibroblasts (CRL1904) | 37 μg/mL (187µM) | 1.5 μg/mL (7.6µM) |
| Microvascular endothelial cells (HMEC-1) | 0.48μg/mL (2.4µM) | 0.19 μg/mL (0.95µM) |
| Monocytes (TIB202) | 50μg/mL (252.7µM) | 6 μg/mL (30µM) |
Cytotoxicity Assay

Cell Proliferation Assay

Discussion
Acknowledgments
References
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxfacts for Arsenic (Update); U.S. Department of Health and Human Services, Public Health Service: Atlanta, GA, 2000. online at http://www.atsdr.cdc.gov/tfacts2.html.
- U.S. Environmental Protection Agency. Integrated Risk Information System (IRIS) on Arsine; Environmental Criteria and Assessment Office, Office of Health and Environmental Assessment, Office of Research and Development: Cincinnati, OH, 1993.
- World Health Organization (WHO). Arsenic in Drinking Water. Fact sheet No 210; Geneva, Switzerland, 2001. [Google Scholar]
- Gandolfi, A. J. Molecular Effects of Low Level Exposure to Arsenic. 2000; [email protected]. [Google Scholar]
- Quinby, G. Melanocytes and Melanogenesis. Case Western University, 2000. On line at: http://dermed.cwru.edu/residents/melanocyte.htm.
- Waclavicek, M.; Berer, A.; Oehler, L.; Stockl, J.; Schloegl, E.; Majdic, O.; Knapp, W. Calcium ionophore: a single reagent for the differentiation of primary human acute myelogenous leukaemia cells towards dendritic cells. Br. J. Haematol. 2001, 114(2), 466–73. [Google Scholar] [CrossRef]
- Ali, A.; Lock, J. Physiological and toxicological changes in the skin resulting from the action and interaction of metal ions. Crit. Rev. Toxicol. 1995, 25, 397–462. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M. C.; Yih, L. H.; Haung, R. N.; Peck, K.; Lee, T. C. Tumor formation of immortalized HaCaT cells in nude mice by long term exposure to sodium arsenite at non toxic doses. Toxicology 2001, 164, 95. [Google Scholar]
- Chen, C-S. J.; Siegel, D. M. Arsenical keratosis. eMedicine J. 2001, 2(6). [Google Scholar]
- Bae, D. S.; Gennings, C.; Carter, W. H., Jr.; Yang, R. S.; Campain, J. A. Toxicological interactions among arsenic, cadmium, chromium, and lead in human keratinocytes. Toxicol. Sci. 2001, 63, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Germolec, D.R.; Yoshida, T.; Gaido, K.; Wilmer, J. L.; Simeonova, P. P.; Kayama, F.; Burleson, F.; Dong, W.; Lange, R. W.; Luster, M. I. Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol. Appl. Pharmacol. 1996, 141, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Vega, L.; Styblo, M; Patterson, R.; Cullen, W.; Wang, C.; Germolec, D. Differential effects of trivalent and pentavalent arsenicals on cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol. Appl. Pharmacol. 2001, 172(3), 225–232. [Google Scholar]
- Rojewski, M. T.; Baldus, C.; Knauf, W.; Thiel, E.; Schrezenmeier, H. Dual effects of arsenic trioxide (As2O3) on non-acute promyelocytic leukaemia myeloid cell lines: induction of apoptosis and inhibition of proliferation. Br. J. Haematol. 2002, 116, 555–563. [Google Scholar]
- Trouba, K. J.; Glanzer, J. G.; Vorce, R. L. Wild-type and Ras-transformed fibroblasts display differential mitogenic responses to transient sodium arsenite exposure. Toxicol. Sci. 1999, 50, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. Q.; Zhu, J.; Shi, X. G.; Ni, J. H.; Zhong, H. J.; Si, G. Y.; Jin, X. L.; Tang, W.; Li, X. S.; Xong, S. M.; Shen, Z. X.; Sun, G. L.; Ma, J.; Zhang, P.; Zhang, T. D.; Gazin, C.; Naoe, T.; Chen, S. J.; Wang, Z. Y.; Chen, Z. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar]
- Jing, Y.; Dai, J.; Chalmers-Redman, R. M.; Tatton, W. G.; Waxman, S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 1999, 94, 2102–2111. [Google Scholar] [PubMed]
- Jia, P.; Chen, G.; Huang, X.; Cai, X.; Yang, J.; Wang, L.; Zhou, Y.; Shen, Y.; Zhou, L.; Yu, Y.; Chen, S.; Zhang, X.; Wang, Z. Arsenic trioxide induces multiple myeloma cell apoptosis via disruption of mitochondrial transmembrane potentials and activation of caspase-3. Chin. Med. J. (Engl) 2001, 114, 19–24. [Google Scholar]
- Vogt, B.; Rossman, T. Effects of arsenite on p53, p21 and cyclin D expression in normal human fibroblasts a possible mechanism for arsenite's comutagenicity. Mutat. Res. 2001, 478, 159–168. [Google Scholar] [CrossRef] [PubMed]
© 2003 by MDPI (http://www.mdpi.org).
Share and Cite
Graham-Evans, B.; Tchounwou, P.B.; Cohly, H.H.P. Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells. Int. J. Mol. Sci. 2003, 4, 13-21. https://0-doi-org.brum.beds.ac.uk/10.3390/i4010013
Graham-Evans B, Tchounwou PB, Cohly HHP. Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells. International Journal of Molecular Sciences. 2003; 4(1):13-21. https://0-doi-org.brum.beds.ac.uk/10.3390/i4010013
Chicago/Turabian StyleGraham-Evans, Barbara, Paul B. Tchounwou, and Hari H. P. Cohly. 2003. "Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells" International Journal of Molecular Sciences 4, no. 1: 13-21. https://0-doi-org.brum.beds.ac.uk/10.3390/i4010013