
4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline


1
Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
2
Center for Drug Design, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455, USA
*
Author to whom correspondence should be addressed.
Molbank 2021, 2021(4), M1305; https://0-doi-org.brum.beds.ac.uk/10.3390/M1305
Submission received: 21 October 2021
/
Revised: 1 December 2021
/
Accepted: 2 December 2021
/
Published: 9 December 2021
(This article belongs to the Special Issue Quinoline, Derivatives and Applications)
Abstract
:4-Amino-imidazo-, oxazolo-, and thiazoloquinolines are key structural scaffolds in the design of nucleoside base analogs for use as therapeutic agents. Current strategies for arriving at diverse substitutions at the C6–C9 positions of the thiazolo- and oxazoloquinolines, however, are limited due to difficulties in arriving at the thiazoloquinoline-5N-oxide intermediate using electron deficient aromatic systems. Here, we demonstrate a synthetic route to obtain substituted thiazoloquinolines with electron-withdrawing groups at the C7 position. The target compound, 4-amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline, is obtained in eight steps using a 7-bromo surrogate as a precursor to the successful generation of the N-oxide intermediate, and final transformation via Pd-mediated C7-acylation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The quinoline ring system is a key building block in the design of a variety of tricyclic heterocycles including imidazo-, oxazolo- and thiazoloquinolines. These molecular scaffolds have found great utility in the design of nucleoside base analogs that activate the innate immune system through Toll-like receptors (TLR) 7 and 8 [1,2,3,4,5,6,7]. When triggered, these receptors stimulate the release of proinflammatory cytokines that are critical to fighting cancer and infectious diseases. Substitutions to the heterocycle have been shown to correlate with both selectivity and potency in driving TLR7 and TLR8 signaling, and more desirable cytokine skewing in the design of adjuvants and immunostimulants for treating cancer [5,6]. Historically, compounds based on the imidazoquinoline scaffold, such as imiquimod (1), were synthesized starting from the appropriately substituted quinoline [3]. These early routes, however, were somewhat limited in versatility due to the use of harsh reaction conditions that prevented the installation of diverse functional groups at the C6–C9 positions. Our group reported an efficient approach to arrive at highly substituted compounds via a Suzuki–Miyaura cross-coupling reaction between iodoimidazoles (1b) and phenylboronic acids (1c) to produce 5-phenylimidazoles (1a) that can be subsequently cyclized to yield the desired tricycle, as shown in Figure 1A [5]. This route, however, is not readily adapted to obtaining oxazolo- or thiazoloquinolines, which are key analogs in the development of potent TLR agonists. The synthesis of these analogs begins with the aptly substituted quinoline (2c), which is transformed into 4-aminothiazoloquinoline through the N-oxide derivative (2a) of 2b [4]. In our attempts to diversify the C6–C9 positions of CL075 (a commercially available TLR7/8 standard), this reaction sequence failed to yield the desired 5N-oxide intermediate in the presence of an electron-withdrawing substituent at C7. This limitation may, in part, explain the lack of examples in the literature with diverse functional substitutions at these positions.
The ability to strategically substitute thiazoloquinolines (such as CL075) is critical to establishing the limits of activity and utility of this scaffold as a nucleoside base mimic or as a TLR7 and TLR8 agonist. Prior work has shown that the addition of an ester to the C7 position of 4-amino-imidazoquinolines dramatically impacts the activity of these compounds [6]. Our initial attempts to install an ester to the 4-amino-thiazoloquinoline scaffold (2) followed the existing synthetic route outlined in Figure 1B. As noted above, this approach failed to yield the 5N-oxide intermediate which we attribute to the inductive effect of the acyl group in the C7 position (as depicted in Figure 2). Given the lack of literature precedence to support the standard pathway in the presence of electron-withdrawing groups, we developed an alternative route to functionalize these compounds that utilizes palladium-catalyzed alkoxy carbonylation to acylate the C7 position [8].
2. Results and Discussion
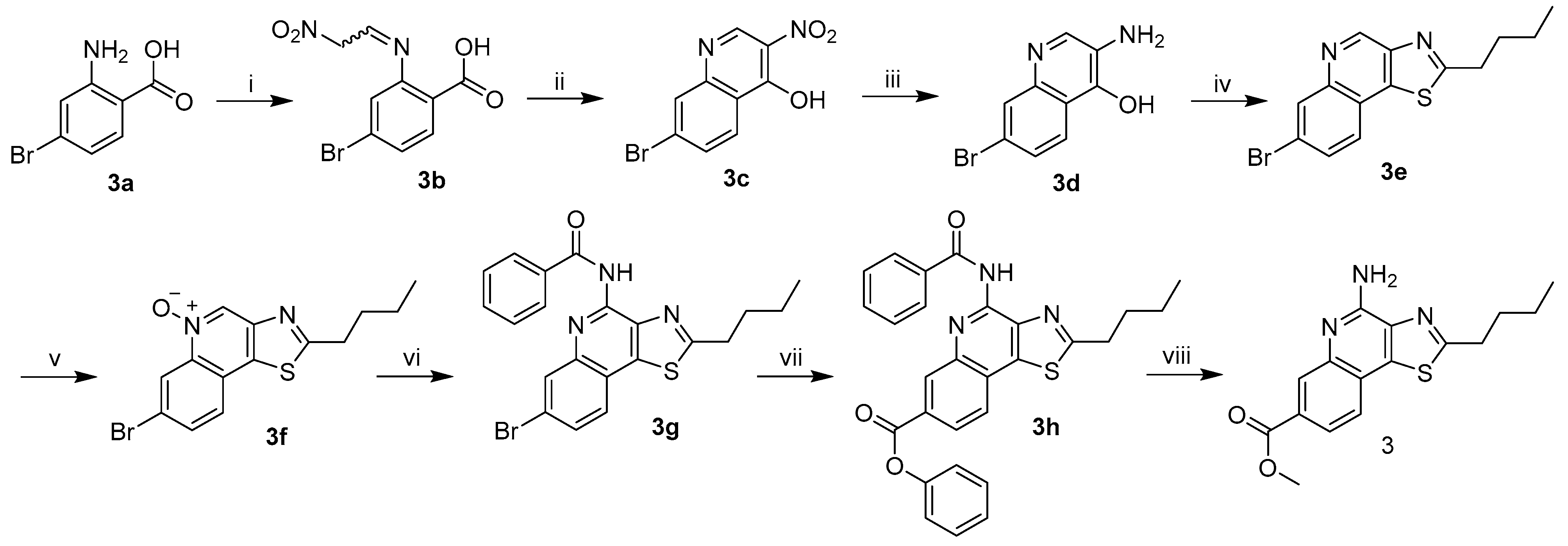
The procedure described in Scheme 1 begins with the installation of a bromo group into thiazoloquinoline using a modified Niementowski reaction to produce the desired 3-amino-4-hydroxy-7-bromoquinoline (3d) [9,10]. Cyclization to the 7-bromo-thiazoloquinoline (3e) was achieved by first forming the primary amide at the 3-amino position, followed by dehydration using phosphorous pentasulfide in pyridine. Oxidation of the N5 position using 3-chloroperbenzoic acid in dichloromethane in the presence of the C7-bromo group proceeded smoothly and the N5-oxide (3f) was obtained in 93% yield (with no evidence of the N3 oxidation product). The weaker deactivating effect of the halogen, as compared with the acyl group, was easily overcome in this case by using additional equivalents (3.5) of the peroxide. The 4-amino group was installed as a benzamido surrogate via a 1,3-dipolar cycloaddition reaction using dipolarophile benzoyl isocyanate, with carbon dioxide being released as a byproduct. The final product was obtained in a two-step sequence that generates the C7-phenyl ester using Pd2(dba)3 with XantPhos as the catalyst for alkoxycarbonylation, followed by acid-catalyzed hydrolysis and transesterification in methanol to yield the target compound 3 in 10.8% yield.
3. Materials and Methods
General Experimental Conditions and Materials: Bulk solvents were purchased from Fisher Chemical and Macron; general chemicals were purchased from Acros Organics, Alfa Aesar, Fisher Chemical, Macron, Mallinckrodt Specialty Chemicals, Oakwood Chemical, Sigma-Aldrich, and TCI America and were used as received. SiliaFlash P60 silica gel was purchased from Silicycle. Nitromethane was purchased from Alfa Aesar. 4-Bromoanthranilic acid was purchased from Oakwood Chemical. Sodium dithionite was purchased from J.T. Baker. Valeryl chloride and 9,9-dimethyl-9H-xanthene-4,5-diylbis(diphenylphosphane) (XantPhos) were purchased from TCI America. Benzoyl isocyanate and phenol were purchased from Sigma-Aldrich. Tris(dibenzylideneacetone)dipalladium was purchased from Chem-Impex. An anhydrous solvent dispensing system (MBraun MB-SPS-800 or Inert PureSolv MD 5) was used for drying toluene and this solvent was dispensed under nitrogen. Thin-layer chromatography (TLC) was performed on 0.25 mm hard-layer silica GF plates. Developed plates were visualized with a hand-held UV lamp. Column chromatography conditions were based on TLC results. Some 1H NMR spectra were recorded on a Varian 400 MHz spectrometer with a dual broadband probe, while some 1H NMR and 13C{1H} NMR spectra were recorded on a Bruker 600 MHz Avance NEO spectrometer with a 5 mm triple resonance cryoprobe. Proton chemical shifts are reported in ppm from internal standards of residual chloroform (CDCl3-7.26 ppm) or dimethylsulfoxide ((CD3)2SO-2.50 ppm). 13C{1H} chemical shifts are reported in ppm from internal standards of residual chloroform (CDCl3-77.0 ppm) or dimethylsulfoxide ((CD3)2SO-39.5 ppm). Proton chemical data are reported as follows: chemical shift (multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet, br = broad, prefixed “app-” = apparent), coupling constant, and integration). High-resolution mass spectra were obtained on an Agilent TOF II TOF/MS instrument, equipped with an ESI or APCI interface or on a Bruker BioTOF II with an ESI interface. (See Supplementary Materials for 1H NMR, 13C{1H} NMR, and HRMS spectra.)
The preparation of methazoic acid was adapted from established methods [10]. Sodium hydroxide (6.72 g, 168 mmol) was dissolved in water (12 mL) and maintained at 25 °C. To this aqueous sodium hydroxide, nitromethane (3 mL, 56 mmol) was added dropwise with rapid stirring and heated to 35 °C for 10 min. The reaction was cooled to 25 °C and another portion of nitromethane (3 mL, 56 mmol) was added while carefully maintaining the temperature at or below 35 °C. After 10 min of rapid stirring, the reaction was heated to 50 °C for 5 min and then allowed to cool to 25 °C over 10 min. The reaction mixture was poured into cold water (50 mL) and concentrated hydrochloric acid (14 mL, 167 mmol) was then added. Additional water was added until the total volume reached 80 mL. The crude methazoic acid solution should be used immediately in the following step or frozen (−20 °C) and carefully thawed just before use. (Note: Although dilute methazoic acid or sodium methazoate is typically not a shock or temperature sensitive explosive, methazoic acid containing salts or solutions can be explosive).
4-Bromo-2-((2-nitroethylidene)amino)benzoic acid (3b). To a fully dissolved solution of 4-bromoanthranilic acid (3.0 g, 13.89 mmol, 1 equiv) in a 1:1 mixture of water:dioxane (90 mL) and concentrated hydrochloric acid (1.2 mL, 14 mmol, ~1 equiv) at 50 °C, was added the crude aqueous methazoic acid solution (40 mL, ~28 mmol, ~2 equiv), freshly prepared as described above. An off-white precipitate immediately began to form and the reaction was allowed to cool to 25 °C and stirred for 18 h. The pale-yellow precipitate was filtered and rinsed with water until the filtrate was clear. The filtered solid was dried to constant mass (3.1 g, 78%) in a vacuum oven (50 °C, ~30 mbar). This can be recrystallized from 45 mL of 1:2 EtOAc:MeOH / gram of crude product to reduce the presence of impurities in later intermediates, but recrystallization did not have a consistent effect on the yield of the next reaction. The recrystallized solid has a stronger yellow color. The purity was qualitatively shown to have improved by TLC. The solid was carried forward without additional characterization: Rf = 0.15 (97:2:1 EtOAc:MeOH:H2O).
7-Bromo-3-nitroquinolin-4-ol (3c). 3b (0.65 g, 2.28 mmol, 1 equiv) was added to a flask with stir bar and acetic anhydride (7.5 mL). This suspension was heated to 100 °C for 45 min or until fully dissolved. Anhydrous sodium acetate (0.51 g, 6.27 mmol, 2.75 equiv) was added to the reaction in one portion while vigorously stirring. The reaction was heated to reflux for 1 h and then allowed to cool to 25 °C. The contents of the flask were diluted with cooled 80% aqueous acetic acid and the solid was filtered. The solid was rinsed with acetic acid until the filtrate was clear. The solid was dried in a vacuum oven (50 °C, ~30 mbar) for 18 h to give a tan powder (0.35 g, 57%) that easily lifted off of filter paper. The crude solid exhibited poor solubility in most solvents and was carried on without characterization.
3-Amino-7-bromoquinolin-4-ol (3d). To a solution of crude 3c (0.22 g, 0.82 mmol, 1 equiv) in anhydrous DMF (15 mL) was added sodium dithionite (1.15 g, 6.6 mmol, 8 equiv). The reaction mixture was stirred for 18 h under N2 at 25 °C. DMF was removed in vacuo and the crude solid was suspended in ethyl acetate (20 mL) and then washed with hot water (20 mL). The organic layer was clear and the aqueous layer was dark brown. This mixture was stirred and heated to 70 °C while solid sodium carbonate powder (2.5 g) was added and allowed to fully dissolve. The organics (now brown) and solids at the interface were separated from the aqueous layer (a lighter yellowish brown). The aqueous layer was carefully extracted with ethyl acetate (4 × 10 mL). The product exhibited reasonable solubility in the aqueous layer. Organics (with solids) were combined and concentrated in vacuo. The crude compound was purified by silica column 85:9:3:3 EtOAc:MeOH:H2O:Et3N to yield 0.09 g (46%) of 3d as an off-white solid: 1H NMR ((CD3)2SO, 400 MHz) δ 11.46 (br s, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.67 (s, 1H), 7.53 (s, 1H), 7.28 (d, J = 8.8 Hz, 1H), 4.53 (br s, 2H). 13C{1H} NMR ((CD3)2SO, 600 MHz) δ 169.2, 138.0, 131.9, 127.1, 124.1, 122.8, 120.3, 120.0, 119.3. (ESI+): Calcd C9H8BrN2O [M + H] + 238.9815, found 238.9810 (error 1.8 ppm).
7-Bromo-2-butylthiazolo[4,5-c]quinoline (3e). To a solution of 3d (0.07 g, 0.29 mmol, 1 equiv) in anhydrous pyridine (3.4 mL), being purged with N2, was added valeryl chloride (0.044 g, 0.37 mmol, 1.25 equiv). This mixture was heated to 60 °C for 2 h in a pressure vessel. The reaction was cooled to 25 °C and again placed under a stream of N2. Phosphorus pentasulfide (0.14 g, 0.32 mmol, 1.1 equiv) was added and the reaction was heated to 105 °C for 18 h in the pressure vessel. The reaction was cooled to 25 °C and aqueous sodium carbonate (10 mL) and ethyl acetate (10 mL) were added. The mixture was separated and the aqueous layer extracted with ethyl acetate (2 × 5 mL). (Caution: there will be trace hydrogen sulfide in the organics. Pulling a light vacuum with some gentle heating for 2 h will usually remove it). The organics were combined and concentrated in vacuo and purified by silica column 1:99 MeOH:DCM to give 0.08 g (86%) of 3e as a yellow waxy solid: 1H NMR (CDCl3, 400 MHz) δ 9.43 (s, 1H), 8.43 (d, J = 1.6 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.73 (dd, J = 8.4, 1.6 Hz, 1H), 3.23 (t, J = 7.6 Hz, 2H), 1.94 (quint, J = 7.6 Hz, 2H), 1.51 (app-sext, J = 7.6, 7.2 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 173.3, 147.9, 146.5, 144.7, 140.5, 132.8, 130.8, 126.0, 122.4, 122.1, 34.0, 31.7, 22.2, 13.7. (ESI+): Calcd C14H14BrN2S [M + H] + 321.0056, found 321.0068 (error 3.8 ppm).
7-Bromo-2-butylthiazolo[4,5-c]quinoline 5-oxide (3f). To a solution of 3e (0.075 g, 0.23 mmol, 1 equiv) in dichloromethane (3 mL) was added 53% m-chloroperoxybenzoic acid (0.27 g, 0.82 mmol, 3.5 equiv). This was stirred at 25 °C for 15 h. TLC indicated a near-complete reaction. The reaction mixture was washed with saturated aqueous sodium carbonate (10 mL) and diluted with an additional portion of dichloromethane (10 mL) and water (10 mL) before the mixture was separated, the aqueous layer was extracted with DCM (2 × 10 mL), and the organics were combined and concentrated in vacuo. The crude solid 3f was purified by silica column 1:99–2:98 MeOH:DCM (linear gradient) to give 0.073 g (93%) of a white powder: 1H NMR (CDCl3, 400 MHz) δ 9.19 (s, 1H), 9.11 (s, 1H), 7.84 (s, 2H), 3.20 (t, J = 7.6 Hz, 2H), 1.92 (quint, J = 7.6 Hz, 2H), 1.51 (sext, J = 7.6 Hz, 2H), 1.00 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 175.6, 147.2, 139.2, 133.3, 132.1, 130.6, 126.7, 124.3, 124.0, 122.5, 34.1, 31.7, 22.2, 13.7. (ESI+): Calcd C14H13BrN2OSNa [M + Na] + 358.9824, found 358.9809 (error 4.4 ppm).
N-(7-Bromo-2-butylthiazolo[4,5-c]quinolin-4-yl)benzamide (3g). To a solution of 3f (0.073 g, 0.22 mmol, 1 equiv) in dichloromethane (8 mL) was added benzoyl isocyanate (0.127 g, 0.86 mmol, 4 equiv). The solution turned a light-yellow color after the benzoyl isocyanate was fully dissolved and was stirred at 25 °C for 18 h. The reaction mixture was concentrated in vacuo and purified by silica column 0:100–1:99 MeOH:DCM (linear gradient) which resulted in 0.070 g (73%) of 3g as a white solid: 1H NMR (CDCl3, 400 MHz) δ 9.69 (br s, 1H), 8.41 (s, 1H), 8.08 (s, 1H), 8.06 (s, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.65–7.53 (m, 4H), 3.21 (t, J = 7.6 Hz, 2H), 1.92 (quint, J = 7.6 Hz, 2H), 1.53 (app-sext, J = 7.6, 7.2 Hz, 2H), 1.02 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 173.0, 164.6, 145.4, 144.3, 140.7, 139.3, 134.7, 132.4, 132.2, 129.2, 128.9, 127.7, 125.5, 122.9, 120.2, 34.0, 31.9, 22.3, 13.8. (ESI+): Calcd C21H19BrN3OS [M + H] + 440.0427, found 440.0437 (error 2.3 ppm).
N-(2-Butyl-7-phenoxycarbonylthiazolo[4,5-c]quinolin-4-yl)benzamide (3h). A solution of formic acid (2 mL) in acetic anhydride (5 mL) was stirred at 35 °C for 1 h to generate acetic formic anhydride. 3g (0.07 g, 0.16 mmol, 1 equiv), phenol (0.061 g, 0.65 mmol, 4.1 equiv), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (0.015 g, 0.016 mmol, 10 mol%) and 9,9-dimethyl-9H-xanthene-4,5-diylbis(diphenylphosphane) (XantPhos) (0.021 g, 0.037 mmol, 23 mol%) were added to a flask and suspended in toluene (4 mL) under a stream of N2. This mixture was stirred for 1 min before the premade acetic formic anhydride (75 μL, 0.95 mmol, 6 equiv) was added in a single portion, followed immediately by the addition of triethylamine (100 μL, 0.72 mmol, 4.5 equiv) while stirring. The flask was capped with a septum and stirred for 2 min. An additional portion of triethylamine was then added (100 μL) (1.44 mmol, 9 equiv total) followed by an additional portion of acetic formic anhydride (30 μL) (1.33 mmol, 8.3 equiv total). The reaction was stirred and heated to 85 °C for 18 h then cooled to 25 °C and diluted with ethyl acetate (5 mL). The crude reaction mixture was filtered through a small silica plug, concentrated in vacuo, and purified by silica column 1:99 MeOH:DCM–4:96 MeOH:DCM to give 0.012 g (16%) of an off-white solid: 1H NMR (CDCl3, 600 MHz) δ 9.76 (br s, 1H), 9.17 (s, 1H), 8.25 (dd, J = 8.4, 1.8 Hz, 1H), 8.09 (d, J = 7.8 Hz, 2H), 7.94 (d, J = 8.4 Hz, 1H), 7.65–7.43 (m, 5H), 7.35–7.75 (m, 3H), 3.25 (t, J = 7.8 Hz, 2H), 1.95 (quint, J = 7.8 Hz, 2H), 1.54 (app-sext, J = 7.8, 7.2 Hz, 2H), 1.03 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 174.4, 164.7, 164.4, 150.9, 145.4, 143.1, 140.3, 140.2, 134.5, 132.8, 132.4, 129.9, 129.6, 129.5, 128.9, 127.6, 126.1, 125.9, 124.7, 121.6, 34.0, 31.9, 22.3, 13.7. (ESI+): Calcd C28H24N3O3S [M + H] + 482.1533, found 482.1572 (error 8.1 ppm).
4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline (3). 3h (0.010 g, 0.020 mmol) was added to a small vial and suspended in anhydrous methanol (3 mL). The mixture was sonicated for 1 min to break up any larger solid chunks. Under a stream of N2, concentrated sulfuric acid (3 drips, approximately 125 mg) was added, which quickly dissolved the fine suspended solids. The vial was capped and then stirred and heated at 60 °C for 15 h. The reaction was cooled to 25 °C and neutralized with solid sodium carbonate powder, diluted with ethyl acetate (5 mL) and water (5 mL), and stirred for 10 min. This mixture was concentrated in vacuo and redissolved in the same amount of ethyl acetate and water. The mixture was separated and the aqueous product was extracted with ethyl acetate (3 × 5 mL). The organics were combined and concentrated in vacuo. The crude product was purified by silica column 25:75 EtOAc:Hexs to give 4.5 mg (69%) of the final product 3 as a white waxy solid: 1H NMR (CDCl3, 600 MHz) δ 8.53 (d, J = 1.2 Hz, 1H), 8.03 (dd, J = 8.4, 1.2 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 6.86 (br s, 2H), 3.98 (s, 3H), 3.20 (t, J = 7.8 Hz, 2H), 1.91 (app-quint, J = 7.8, 7.2 Hz, 2H), 1.51 (sext, J = 7.2 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (CDCl3, 600 MHz) δ 174.5, 166.2, 151.0, 141.5, 138.7, 138.1, 131.5, 125.5, 125.2, 124.6, 121.5, 52.6, 33.9, 31.8, 22.2, 13.7. (APCI+): Calcd C16H18N3O2S [M + H] + 316.1114, found 316.1108 (error 1.9 ppm).
4. Conclusions
A route to install more diverse substitutions at the C7 position of 4-amino-thaizoloquinolin was described and applied to obtain 4-amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline. This approach overcomes the limitations of the traditional route, which requires formation of a 5N-oxide intermediate that is not compatible with electron-withdrawing groups at the C7 position. The proposed method, which exploits a Pd-catalyzed carbonylation reaction, should prove useful in the design of both thiazolo- and oxazoloquinoline nucleoside base analogs, as well as highly substituted isoquinolines.
Supplementary Materials
The following are available online, 1H NMR (Figures S1, S3, S5, S7, S9, and S11), 13C NMR (Figures S2, S4, S6, S8, S10, and S12), and HRMS spectra (Figures S13–S18) of compounds 3d, 3e, 3f, 3g, 3h, and 3.
Author Contributions
Conceptualization, D.M.F. and P.G.L.; methodology, D.M.F. and P.G.L.; investigation, D.M.F. and P.G.L.; resources, D.M.F.; data curation, P.G.L.; writing—original draft preparation, D.M.F.; writing—review and editing, D.M.F. and P.G.L.; supervision, D.M.F.; project administration, D.M.F.; funding acquisition, D.M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was in part supported by a research grant from Seagen, Inc., Bothell, WA, USA, to D.M.F. The NMR spectroscopy component of this research was supported by Grant 1S10OD021536 from the National Institute of General Medical Sciences. The contents of this publication are the sole responsibility of the authors and do not necessarily represent the official views of NIGMS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data is in Supporting Materials.
Acknowledgments
We are grateful to the Minnesota NMR center, https://nmr.umn.edu/ (accessed on 1 December 2021), for providing NMR instrumentation. We also thank John Schultz and Mu Yang for their help with characterization and their insights.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gerster, J.F.; Lindstrom, K.J.; Miller, R.L.; Tomai, M.A.; Birmachu, W.; Bomersine, S.N.; Gibson, S.J.; Imbertson, L.M.; Jacobson, J.R.; Knafla, R.T.; et al. Synthesis and structure-activity-relationships of 1H-Imidazo[4,5-c]quinolines that induce interferon production. J. Med. Chem. 2005, 48, 3481–3491. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.; Salunke, D.B.; Sil, D.; Guo, X.; Salyer, A.C.; Hermanson, A.R.; Kumar, M.; Malladi, S.S.; Balakrishna, R.; Thompson, W.H.; et al. Determinants of activity at human Toll-like receptors 7 and 8: Quantitative structure-activity relationship (QSAR) of diverse heterocyclic scaffolds. J. Med. Chem. 2014, 57, 7955–7970. [Google Scholar] [CrossRef] [PubMed]
- Schiaffo, C.E.; Shi, C.; Xiong, Z.; Olin, M.; Ohlfest, J.R.; Aldrich, C.A.; Ferguson, D.M. Structure–Activity Relationship Analysis of Imidazoquinolines with Toll-like Receptors 7 and 8 Selectivity and Enhanced Cytokine Induction. J. Med. Chem. 2014, 57, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.X.Z.; Chittepu, P.; Aldrich, C.C.; Ohlfest, J.R.; Ferguson, D.F. Discovery of human toll-like receptor 7/8 ligand analogs with novel cytokine induction. ACS Med. Chem. Lett. 2012, 3, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Larson, P.; Kucaba, T.A.; Xiong, Z.; Olin, M.; Griffith, T.S.; Ferguson, D.M. Design and Synthesis of N1-Modified Imidazoquinoline Agonists for Selective Activation of Toll-like Receptors 7 and 8. ACS Med. Chem. Lett. 2017, 8, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, C.L.; Jiang, L.B.; Zhang, W.Q.; Wu, X.F. Palladium-catalyzed alkoxycarbonylation of aryl halids with phenols employing formic acid as the CO source. Catal. Sci. Technol. 2016, 6, 3099–3107. [Google Scholar] [CrossRef]
- Niementowski, S.V. Synthesen von Chinazolinverbindungen. J. Prakt. Chem. 1895, 51, 564. [Google Scholar] [CrossRef] [Green Version]
- Bachman, G.B.; Welton, D.E.; Jenkins, G.L.; Christian, J.E. Quinoline Derivatives from 3-nitro-4-hydroxyquinoline. J. Am. Chem. Soc. 1947, 69, 365–371. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Synthetic strategies to arrive at the 4-amino imidazoquinolines (A) and thiazoloquinolines (B).
Figure 1.
Synthetic strategies to arrive at the 4-amino imidazoquinolines (A) and thiazoloquinolines (B).

Figure 2.
Deactivation of N5.

Scheme 1.
Synthesis of 4-amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline (3). Reagents and conditions: (i) methazoic acid, H2O-dioxane, 40 °C to rt, 15 h, 78%; (ii) sodium acetate, Ac2O, 130 °C, 2 h, 57%; (iii) sodium dithionite, DMF, rt, 15 h, 46%; (iv) (a) C4H9COCl, pyridine, 50 °C, 1 h, (b) P4S10, 110 °C, 15 h, 86%; (v) m-CPBA, DCM, rt, 15 h, 93%; (vi) benzoyl isocyanate, DCM, rt, 15 h, 73%; (vii) (a) Pd2(dba)3, XPhos, PhOH, PhMe, rt, 10 min, (b) HCOOH, Ac2O, (c) NEt3, 90 °C, 15 h, 16%; (viii) conc. H2SO4, MeOH, 65 °C, 15 h, 69%.
Scheme 1.
Synthesis of 4-amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline (3). Reagents and conditions: (i) methazoic acid, H2O-dioxane, 40 °C to rt, 15 h, 78%; (ii) sodium acetate, Ac2O, 130 °C, 2 h, 57%; (iii) sodium dithionite, DMF, rt, 15 h, 46%; (iv) (a) C4H9COCl, pyridine, 50 °C, 1 h, (b) P4S10, 110 °C, 15 h, 86%; (v) m-CPBA, DCM, rt, 15 h, 93%; (vi) benzoyl isocyanate, DCM, rt, 15 h, 73%; (vii) (a) Pd2(dba)3, XPhos, PhOH, PhMe, rt, 10 min, (b) HCOOH, Ac2O, (c) NEt3, 90 °C, 15 h, 16%; (viii) conc. H2SO4, MeOH, 65 °C, 15 h, 69%.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Larson, P.G.; Ferguson, D.M. 4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline. Molbank 2021, 2021, M1305. https://0-doi-org.brum.beds.ac.uk/10.3390/M1305
AMA Style
Larson PG, Ferguson DM. 4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline. Molbank. 2021; 2021(4):M1305. https://0-doi-org.brum.beds.ac.uk/10.3390/M1305
Chicago/Turabian StyleLarson, Peter G., and David M. Ferguson. 2021. "4-Amino-2-butyl-7-methoxycarbonylthiazolo[4,5-c]quinoline" Molbank 2021, no. 4: M1305. https://0-doi-org.brum.beds.ac.uk/10.3390/M1305
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.