
Molecular Characterization of the Common Snook, Centropomus undecimalis (Bloch, 1792) in the Usumacinta Basin

 ,
,
Abstract
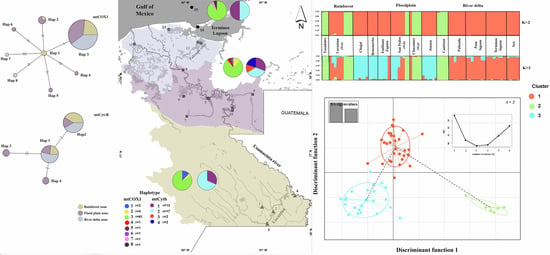
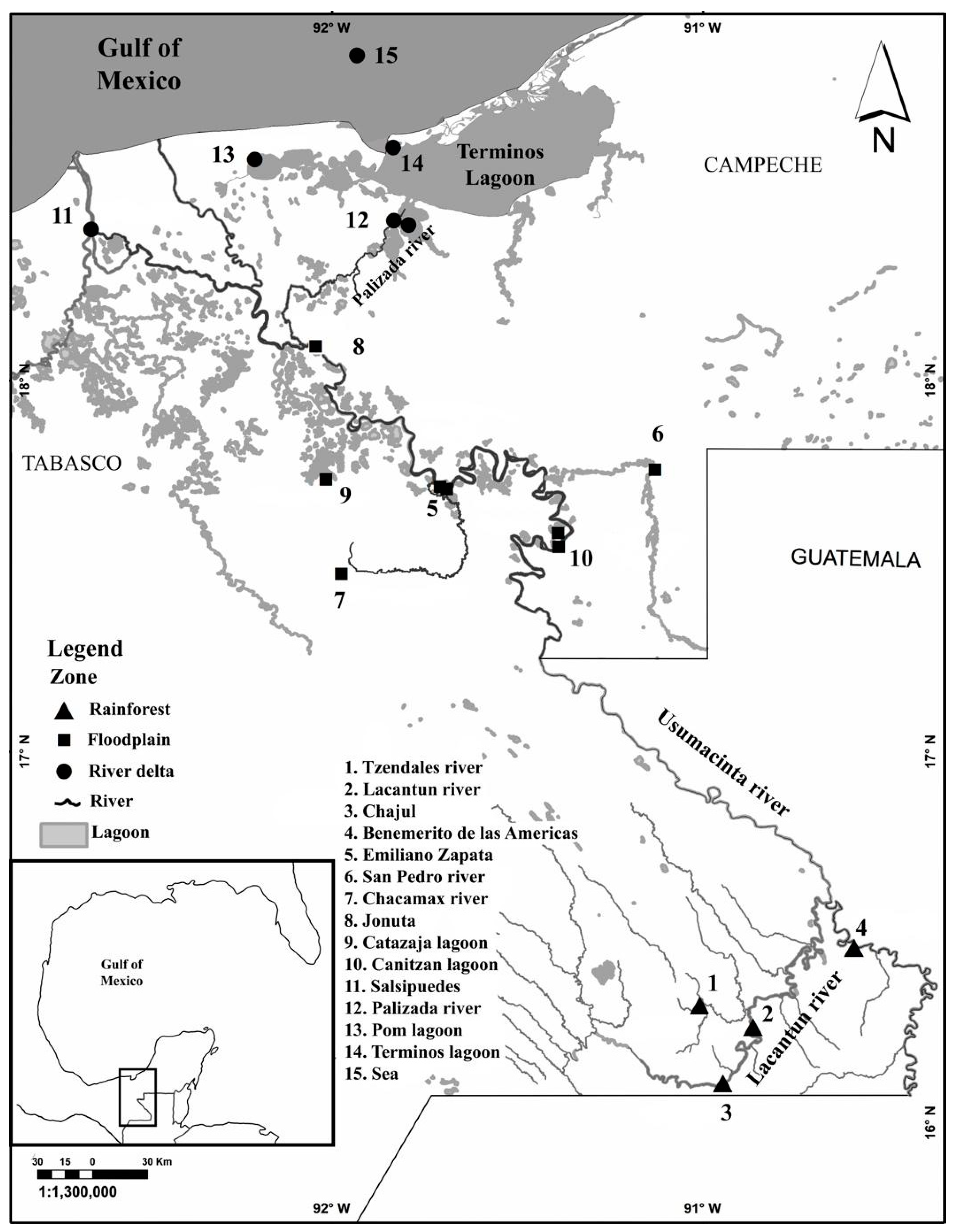
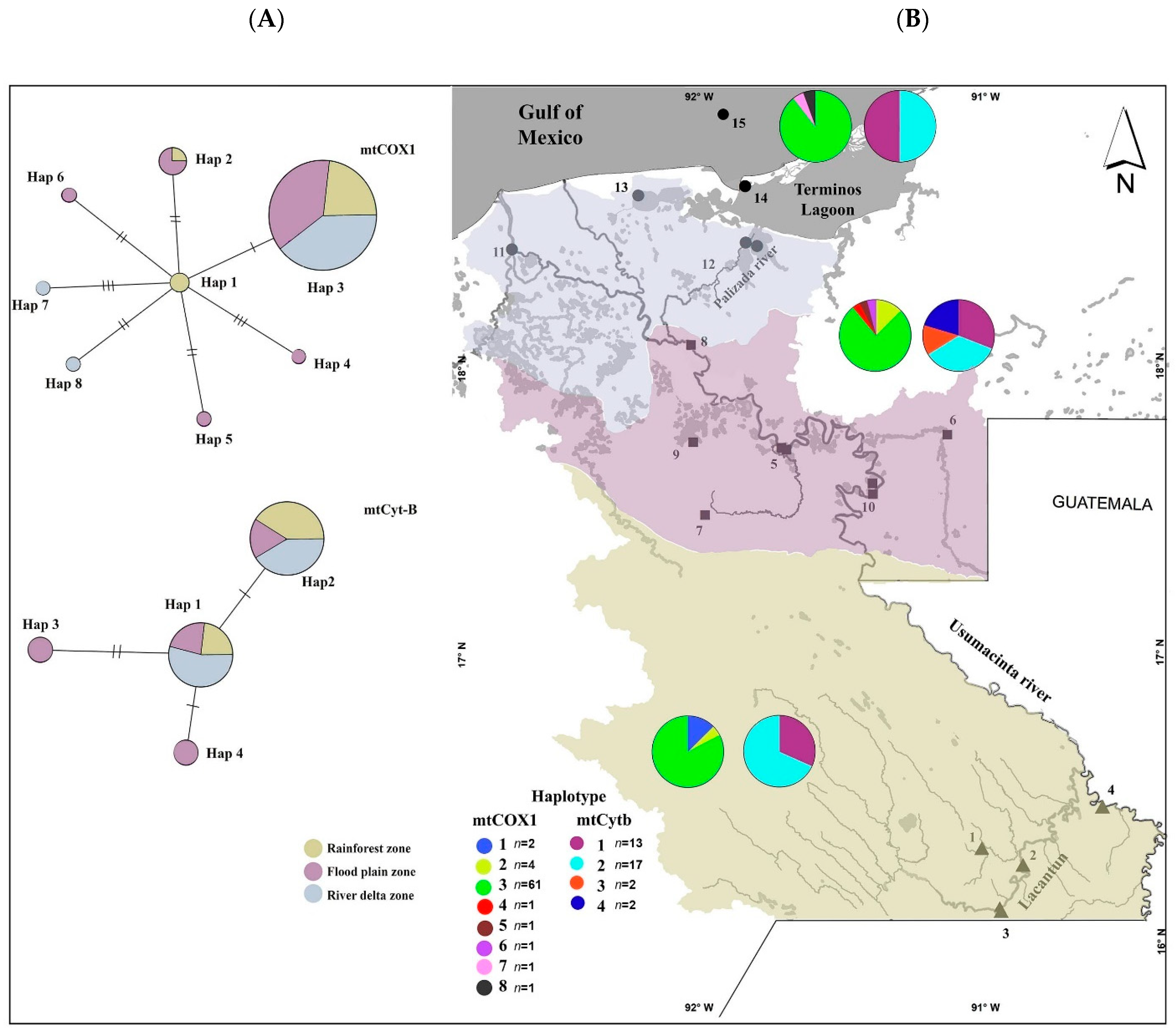
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitochondrial and Nuclear Amplification
2.3. mtDNA Diversity and Genetic Differentiation
2.4. Microsatellite Diversity and Genetic Differentiation
2.5. Isolation by Distance
3. Results
3.1. Microsatellite Structure
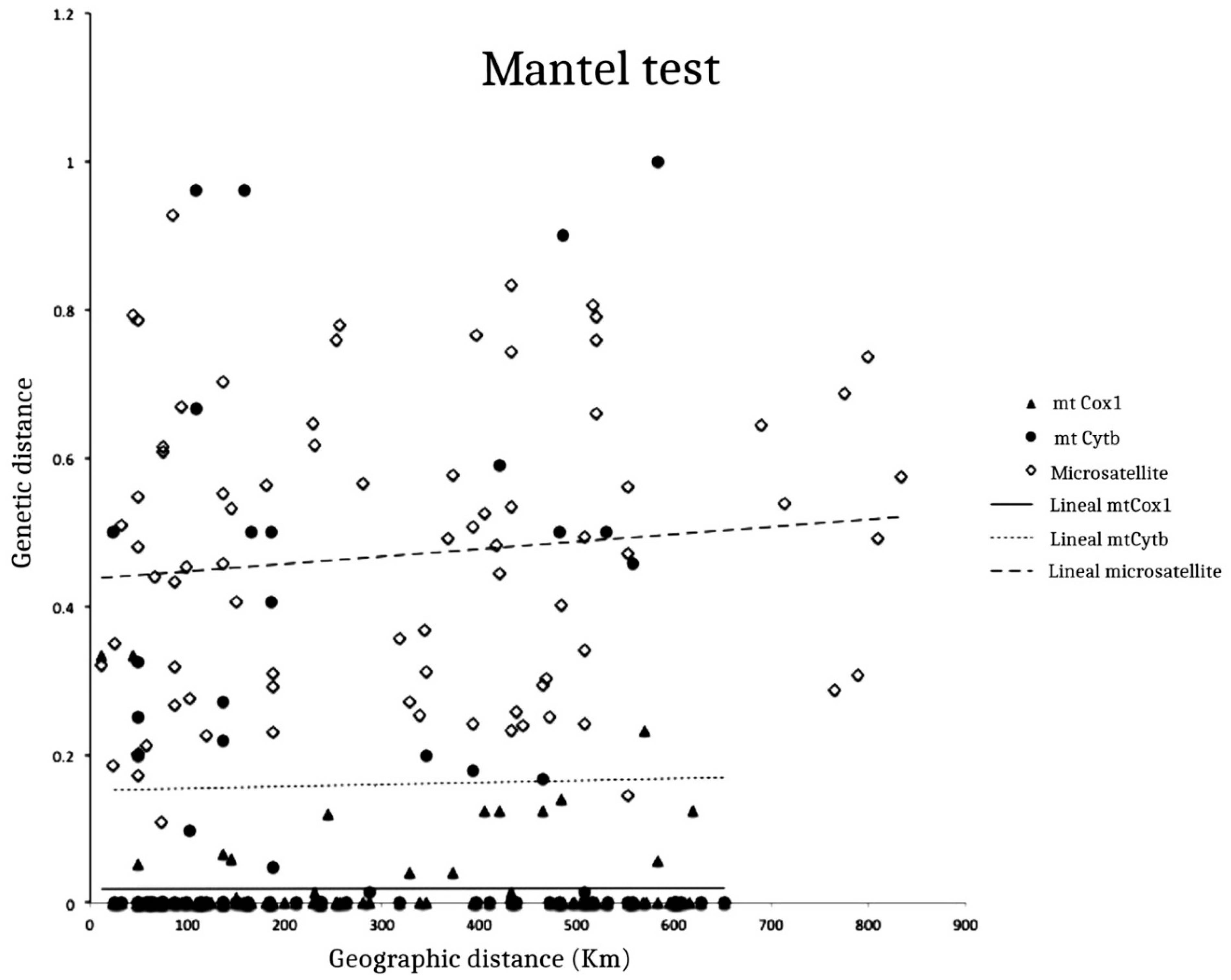
3.2. Isolation by Distance
4. Discussion
4.1. mtDNA Genetic Structure
4.2. nucDNA Genetic Structure
4.3. Gene Flow and Isolation by Distance
4.4. Implications for Species Conservation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pringle, C.M.; Freeman, M.C.; Freeman, B.J. Regional Effects of Hydrologic Alterations on Riverine Macrobiota in the New World: Tropical–Temperate Comparisons. BioScience 2000, 50, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Pringle, C. What Is Hydrologic Connectivity and Why Is It Ecologically Important? Hydrological Process: Hoboken, NJ, USA, 2003; Volume 17, pp. 2685–2689. [Google Scholar]
- Calles, O.; Greenberg, L. Connectivity is a two-way street-the need for a holistic approach to fish passage problems in regulated rivers. River Res. Appl. 2009, 25, 1268–1286. [Google Scholar] [CrossRef]
- Ward, J.V. The Four-Dimensional Nature of Lotic Ecosystems. J. N. Am. Benthol. Soc. 1989, 8, 2–8. [Google Scholar] [CrossRef]
- Branco, P.; Segurado, P.; Santos, J.; Pinheiro, P.; Ferreira, M. Does longitudinal connectivity loss affect the distribution of freshwater fish? Ecol. Eng. 2012, 48, 70–78. [Google Scholar] [CrossRef]
- Lucas, M.C.; Baras, E. Migration of Freshwater Fishes; Lucas, M., Baras, E., Thom, T., Duncan, A., Slavík, O., Eds.; Blackwell Science: Maden, MA, USA, 2001. [Google Scholar]
- Yi, Y.; Yang, Z.; Zhang, S. Ecological influence of dam construction and river-lake connectivity on migration fish habitat in the Yangtze River basin, China. Procedia Environ. Sci. 2010, 2, 1942–1954. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.M.; Bickford, N.; McIntyre, P.B.; Coulon, A.; Ulseth, A.J.; Taphorn, D.C.; Flecker, A.S. Population Structure of a Neotropical Migratory Fish: Contrasting Perspectives from Genetics and Otolith Microchemistry. Trans. Am. Fish. Soc. 2013, 142, 1192–1201. [Google Scholar] [CrossRef]
- Hand, B.K.; Lowe, W.H.; Kovach, R.P.; Muhlfeld, C.C.; Luikart, G. Landscape community genomics: Understanding eco-evolutionary processes in complex environments. Trends Ecol. Evol. 2015, 30, 161–168. [Google Scholar] [CrossRef]
- Garcia, C.P.O.; De Biología, U.I.; Bernal, C.F.M.; Hernández, R.R.; Biomédicas, U.I.D.I. Ecosur Evaluación de la Diversidad de Linajes en Sistemas Dulceacuícolas tropicales (D-LSD): El Sistema Usumacinta como caso de estudio. In Antropización: Primer Análisis Integral; Universidad Nacional Autónoma de México, Centro de Investigaciones en Geografía Ambiental: Mexico City, Mexico, 2019; pp. 125–148. [Google Scholar]
- Oosthuizen, C.J.; Cowley, P.D.; Kyle, S.R.; Bloomer, P. High genetic connectivity among estuarine populations of the riverbream Acanthopagrus vagus along the southern African coast. Estuar. Coast. Shelf Sci. 2016, 183, 82–94. [Google Scholar] [CrossRef]
- Sork, V.L.; Smouse, P.E. Genetic analysis of landscape connectivity in tree populations. Landsc. Ecol. 2006, 21, 821–836. [Google Scholar] [CrossRef]
- DeSalle, R.; Goldstein, P. Review and Interpretation of Trends in DNA Barcoding. Front. Ecol. Evol. 2019, 7, 302. [Google Scholar] [CrossRef] [Green Version]
- Ornelas-García, C.P.; Domínguez-Domínguez, O.; Doadrio, I. Evolutionary history of the fish genus Astyanax Baird & Girard (1854) (Actinopterygii, Characidae) in Mesoamerica reveals multiple morphological homoplasies. BMC Evol. Biol. 2008, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Valdez-Moreno, M.; Ivanova, N.V.; Elías-Gutiérrez, M.; Pedersen, S.L.; Bessonov, K.; Hebert, P.D.N. Using eDNA to biomonitor the fish community in a tropical oligotrophic lake. PLoS ONE 2019, 14, e0215505. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R. Stress and adaptation in conservation genetics. J. Evol. Biol. 2005, 18, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vidal, U.; Chiappa-carrara, X.; Contreras-Sánchez, W.M. Reproductive variability of the common snook, Centropomus undecimalis, in environments of contrasting salinities interconnected by the Grijalva–Usumacinta fluvial system. Cienc. Mar. 2014, 40, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Tringali, M.; Bert, T. The genetic stock structure of common snook (Centropomus undecimalis). Can. J. Fish. Aquat. Sci. 1996, 53, 974–984. [Google Scholar] [CrossRef]
- Mendonça, J.T.; Chao, L.; Albieri, R.J.; Giarrizzo, T.; da Silva, F.M.S.; Castro, M.G.; Brick-Peres, M.; Villwock de Miranda, L.; Vieira, J.P. Centropomus undecimalis. The IUCN Red List of Threatened Species 2019: E.T191835A82665184. 2019. Available online: https://0-dx-doi-org.brum.beds.ac.uk/10.2305/IUCN.UK.2019-2.RLTS.T191835A82665184.en (accessed on 30 May 2021).
- Soria-Barreto, M.; González-Díaz, A.A.; Castillo-Domínguez, A.; Álvarez-Pliego, N.; Rodiles-Hernández, R. Diversity of fish fauna in the Usumacinta Basin, Mexico. Rev. Mex. Biodivers. 2018, 89, 100–117. [Google Scholar]
- Garcia, M.A.P.; Mendoza-Carranza, M.; Contreras-Sánchez, W.; Ferrara, A.; Huerta-Ortiz, M.; Gómez, R.E.H. Comparative age and growth of common snook Centropomus undecimalis (Pisces: Centropomidae) from coastal and riverine areas in Southern Mexico. Rev. Biol. Trop. 2013, 61, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Castro-Aguirre, J.L.; Espinosa-Pérez, H.; Schmitter-Soto, J.J. Ictiofauna Estuarino-Lagunar y Vicaria de México; Editorial Limusa: La Paz, Baja California Sur, Mexico, 1999; ISBN 9681857747. [Google Scholar]
- Adams, A.J.; Wolfe, R.K.; Layman, C.A. Preliminary Examination of How Human-driven Freshwater Flow Alteration Affects Trophic Ecology of Juvenile Snook (Centropomus undecimalis) in Estuarine Creeks. Chesap. Sci. 2009, 32, 819–828. [Google Scholar] [CrossRef]
- Miller, R.R. Peces dulceacuícolas de México; CONABIO, SIMAC, ECOSUR, Desert Fishes of Council: Mexico City, Mexico, 2009. [Google Scholar]
- Hernández-Vidal, U.; Lesher-Gordillo, J.; Contreras-Sánchez, W.M.; Chiappa-Carrara, X. Variabilidad genética del robalo común Centropomus undecimalis (Perciformes: Centropomidae) en ambiente marino y ribereño interconectados. Rev. Biol. Trop. 2014, 62, 627–636. [Google Scholar] [CrossRef] [Green Version]
- González-Porter, G.P.; Hailer, F.; Flores-Villela, O.; García-Anleu, R.; Maldonado, J.E. Patterns of genetic diversity in the critically endangered Central American river turtle: Human influence since the Mayan age? Conserv. Genet. 2011, 12, 1229–1242. [Google Scholar] [CrossRef]
- Gonzalez-Porter, G.P.; Maldonado, J.E.; Flores-Villela, O.; Vogt, R.C.; Janke, A.; Fleischer, R.C.; Hailer, F. Cryptic Population Structuring and the Role of the Isthmus of Tehuantepec as a Gene Flow Barrier in the Critically Endangered Central American River Turtle. PLoS ONE 2013, 8, e71668. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Gómez, J. Sistemática Molecular e Historia Evolutiva de la Familia Dermatemydidae; Universidad de Ciencias y Artes de Chiapas: Tuxtla Gutiérrez, Chiapas, Mexico, 2017. [Google Scholar]
- Elías, D.J.; Mcmahan, C.D.; Matamoros, W.A.; Gómez-González, A.E.; Piller, K.R.; Chakrabarty, P. Scale(s) matter: Deconstructing an area of endemism for Middle American freshwater fishes. J. Biogeogr. 2020, 47, 2483–2501. [Google Scholar] [CrossRef]
- Sonnenberg, R.; Nolte, A.W.; Tautz, D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Front. Zoöl. 2007, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R.; Doadrio, I. Phylogenetic relationships of Iberian cyprinids: Systematic and biogeographical implications. Proc. R. Soc. B Boil. Sci. 1998, 265, 1365–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyoum, S.; Tringali, M.D.; Sullivan, J.G. Isolation and characterization of 27 polymorphic microsatellite loci for the common snook, Centropomus undecimalis. Mol. Ecol. Notes 2005, 5, 924–927. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Series 1999, 41, 95–98. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gatew. Comput. Environ. Workshop (GCE) 2010, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Team, R. A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2006. [Google Scholar]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using Adegenet 2.0.0; Imperial College London, MRC Centre for Outbreak Analysis and Modelling: London, UK, 2015. [Google Scholar]
- Criscuolo, N.G.; Angelini, C. StructuRly: A novel shiny app to produce comprehensive, detailed and interactive plots for population genetic analysis. PLoS ONE 2020, 15, e0229330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.A.; Rannala, B. Bayesian Inference of Recent Migration Rates Using Multilocus Genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimatorv2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2013, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. Comparative methods for the analysis of continuous variables: Geometric interpretations. Evolution 2001, 55, 2143–2160. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 457–462. [Google Scholar] [CrossRef]
- Wright, S. ISOLATION BY DISTANCE. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef]
- Yáñez-Arancibia, A.; Day, J.W.; Currie-Alder, B. Functioning of the Grijalva-Usumacinta River Delta, Mexico: Challenges for Coastal Management. Ocean Yearb. Online 2009, 23, 473–501. [Google Scholar] [CrossRef]
- Mendoza-Carranza, M.; Hoeinghaus, D.J.; Garcia, A.M.; Romero-Rodriguez, Á. Aquatic food webs in mangrove and seagrass habitats of Centla Wetland, a Biosphere Reserve in Southeastern Mexico. Neotrop. Ichthyol. 2010, 8, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, C.; Quintana, Y.; Elías, D.J.; Rodiles-Hernández, R. Peces nativos y pesca artesanal en la cuenca Usumacinta, Guatemala. Rev. Mex. Biodivers. 2018, 89, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Barba-Macías, E.; Juárez-Flores, J.; Trinidad-Ocaña, C.; Sánchez-Martínez, A.D.J.; Mendoza-Carranza, M. Socio-ecological Approach of Two Fishery Resources in the Centla Wetland Biosphere Reserve. In Socio-Ecological Studies in Natural Protected Areas; Springer Science and Business Media LLC: Berlin, Germany, 2020; pp. 627–656. [Google Scholar]
- Mirol, P.M.; Routtu, J.; Hoikkala, A.; Butlin, R.K. Signals of demographic expansion in Drosophila virilis. BMC Evol. Biol. 2008, 8, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, F.; Raymond, M. Testing heterozygote excess and deficiency. Genetics 1995, 140, 1413–1419. [Google Scholar] [CrossRef]
- De Meeûs, T. Revisiting FIS, FST, Wahlund effects, and null alleles. J. Hered. 2018, 109, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Crook, D.A.; Buckle, D.J.; Allsop, Q.; Baldwin, W.; Saunders, T.M.; Kyne, P.M.; Woodhead, J.D.; Maas, R.; Roberts, B.; Douglas, M.M. Use of otolith chemistry and acoustic telemetry to elucidate migratory contingents in barramundi Lates calcarifer. Mar. Freshw. Res. 2017, 68, 1554. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-Gaona, S.; Ramos-Ventura, L.J.; Moreno-Sandoval, F.; Jiménez-Pérez, N. del C.; Haas-Ek, M.A.; Muñiz-Delgado, L.E. Diversidad de flora acuática y ribereña en la cuenca del río Usumacinta, México. Rev. Mex. Biodivers. 2018, 89, 3–44. [Google Scholar]
- Vaca, R.A.; Golicher, D.J.; Rodiles-Hernández, R.; Castillo-Santiago, M. Ángel; Bejarano, M.; Navarrete-Gutiérrez, D.A. Drivers of deforestation in the basin of the Usumacinta River: Inference on process from pattern analysis using generalised additive models. PLoS ONE 2019, 14, e0222908. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, A.; De Oliveira, J.; Soares, C.; Araripe, J. A new species of snook, Centropomus (Teleostei: Centropomidae), from northern South America, with notes on the geographic distribution of other species of the genus. Zootaxa 2019, 4671, 81–92. [Google Scholar] [CrossRef]
- De Oliveira, J.N.; Gomes, G.; Rêgo, P.S.D.; Moreira, S.; Sampaio, I.; Schneider, H.; Araripe, J. Molecular data indicate the presence of a novel species of Centropomus (Centropomidae–Perciformes) in the Western Atlantic. Mol. Phylogenet. Evol. 2014, 77, 275–280. [Google Scholar] [CrossRef]
- Castillo-Domínguez, A.; Barba Macías, E.; Navarrete, A. de J.; Rodiles-Hernández, R.; Jiménez Badillo, M. de L. Ictiofauna de los humedales del río San Pedro, Balancán, Tabasco, México. Rev. Biol. Trop. 2011, 59, 693–708. [Google Scholar] [PubMed]
- Anderson, J.; Williford, D.; González-Barnes, A.; Chapa, C.; Martinez-Andrade, F.; Overath, R.D. Demographic, Taxonomic, and Genetic Characterization of the Snook Species Complex ( Centropomus spp.) along the Leading Edge of Its Range in the Northwestern Gulf of Mexico. N. Am. J. Fish. Manag. 2020, 40, 190–208. [Google Scholar] [CrossRef]
- Rocha-Olivares, A.; Garber, N.M.; Stuck, K.C. High genetic diversity, large inter-oceanic divergence and historical demography of the striped mullet. J. Fish Biol. 2000, 57, 1134–1149. [Google Scholar] [CrossRef]
- Song, C.Y.; Sun, Z.C.; Gao, T.X.; Song, N. Structure Analysis of Mitochondrial DNA Control Region Sequences and its Applications for the Study of Population Genetic Diversity of Acanthogobius ommaturus. Russ. J. Mar. Biol. 2020, 46, 292–301. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mtDNA | Locus | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | mtCox | Cyt-b | Cun20 | Cun 10A | Cun 02 | Cun 21A | Cun 04A | Cun 01 | Cun 021B | Cun 06 | Cun 14 | |||
| Rainforest zone n = 23 | n | 17 | 10 | n | 20 | 20 | 21 | 22 | 18 | 23 | 22 | 22 | 22 | |
| h | 3 | 2 | Na | 14.000 | 15.000 | 8.000 | 5.000 | 5.000 | 8.000 | 5.000 | 7.000 | 10.000 | ||
| Hd | 0.323 | 0.466 | Ne | 7.619 | 11.429 | 6.300 | 1.770 | 3.927 | 3.348 | 3.796 | 4.990 | 5.378 | ||
| S | 2 | 1 | Ho | 0.950 | 0.900 | 0.571 | 0.500 | 0.889 | 0.739 | 0.864 | 0.636 | 0.864 | ||
| k | 0.338 | 0.001 | He | 0.869 | 0.913 | 0.841 | 0.435 | 0.745 | 0.701 | 0.737 | 0.800 | 0.814 | ||
| π | 0.0005 | 0.466 | FIS | −0.094 * | 0.014 | 0.321 | −0.150 | −0.193 | −0.054 | −0.173 | 0.204 * | −0.061 | All locus FIS −0.011 | |
| Floodplain zone n = 29 | n | 29 | 10 | n | 24 | 27 | 29 | 29 | 23 | 27 | 29 | 26 | 29 | |
| h | 5 | 4 | Na | 15.000 | 19.000 | 8.000 | 3.000 | 6.000 | 11.000 | 6.000 | 6.000 | 10.000 | ||
| Hd | 0.369 | 0.822 | Ne | 6.776 | 12.678 | 6.570 | 1.597 | 3.792 | 3.455 | 3.103 | 4.711 | 6.029 | ||
| S | 3 | 2 | Ho | 0.708 | 0.815 | 0.448 | 0.483 | 0.696 | 0.556 | 0.724 | 0.577 | 0.862 | ||
| k | 0.458 | 1.08 | He | 0.852 | 0.921 | 0.848 | 0.374 | 0.736 | 0.711 | 0.678 | 0.788 | 0.834 | ||
| π | 0.0008 | 0.002 | FIS | 0.169 | 0.115 | 0.471 * | −0.291 | 0.055 | 0.218 * | −0.068 | 0.268 * | −0.033 * | All locus FIS 0.085 | |
| River delta n = 29 | n | 26 | 14 | n | 29 | 29 | 29 | 29 | 29 | 29 | 29 | 28 | 29 | |
| h | 3 | 2 | Na | 6.000 | 17.000 | 3.000 | 3.000 | 5.000 | 11.000 | 6.000 | 4.000 | 10.000 | ||
| Hd | 0.150 | 0.538 | Ne | 4.890 | 7.410 | 2.683 | 1.279 | 3.266 | 2.448 | 4.102 | 1.744 | 6.207 | ||
| S | 3 | 1 | Ho | 0.690 | 0.862 | 0.552 | 0.241 | 0.621 | 0.586 | 0.793 | 0.571 | 0.759 | ||
| k | 0.230 | 0.538 | He | 0.795 | 0.865 | 0.627 | 0.218 | 0.694 | 0.592 | 0.756 | 0.427 | 0.839 | ||
| π | 0.0004 | 0.001 | FIS | 0.133 | 0.003 | 0.120 | −0.106 | 0.105 | 0.009 * | −0.049 | −0.339 | 0.096 | All locus FIS 0.011 | |
| All | n | 72 | 34 | n | 73 | 76 | 79 | 80 | 70 | 79 | 80 | 76 | 80 | |
| h | 8 | 4 | Na | 18 | 21 | 8 | 6 | 6 | 14 | 6 | 9 | 14 | ||
| Hd | 0.281 | 0.615 | Ne | 4.216 | 5.933 | 3.250 | 1.562 | 3.154 | 2.937 | 3.111 | 2.370 | 4.390 | ||
| S | 7 | 2 | Ho | 0.784 | 0.868 | 0.495 | 0.410 | 0.732 | 0.652 | 0.772 | 0.568 | 0.822 | ||
| k | 0.354 | 0.722 | He | 0.743 | 0.815 | 0.647 | 0.276 | 0.674 | 0.635 | 0.661 | 0.527 | 0.756 | ||
| π | 0.0006 | 0.001 | FIS | 0.071 | 0.049 | 0.331 | −0.203 | −0.015 | 0.067 | −0.097 | 0.113 | 0.001 | ||
| Marker | Variance Among Groups (%) | Variance among Populations within Groups (%) | Variance within Populations (%) | Among Groups FCT/Φ CT | Among Populations FSC/Φ SC | Within Populations FST/Φ ST |
|---|---|---|---|---|---|---|
| mtCox1 | 3.29 | 16.00 | 86.61 | 0.03295 | −0.08010 | −0.04451 |
| mtCytb | 6.13 | 0.88 | 92.99 | 0.06134 | 0.00936 | 0.07013 |
| Microsatellite K = 3 DACP | 11.11 | −0.69 | 89.57 | 0.111 * | −0.007 | 0.104 * |
| Microsatellite K = 2 STRUCTURE | 5.79010 | 3.17138 | 91.03 | 0.0579 | 0.0336 | 0.089 |
| K = 3 STRUCTURE | 1.69 | 4.27 | 94.04 | 0.016 * | 0.043 * | 0.059 * |
| Population | Na | Na Freq. ≥ 5% | Ne | Np |
|---|---|---|---|---|
| Tzendales River | 3.556 | 3.556 | 3.115 | 1 |
| Lacantun River | 5.889 | 5.889 | 4.236 | 4 |
| Chajul | 5.111 | 5.111 | 4.012 | 4 |
| Benemerito | 3.667 | 3.667 | 2.889 | 1 |
| Emiliano Zapata | 4.333 | 4.333 | 3.426 | 2 |
| San Pedro River | 5.333 | 5.333 | 4.082 | 1 |
| Chacamax River | 3.778 | 3.778 | 3.165 | 2 |
| Jonuta | 4.000 | 4.000 | 3.421 | 0 |
| Canitzan Lagoon | 5.222 | 5.222 | 4.016 | 4 |
| Pom Lagoon | 5.111 | 4.000 | 3.415 | 1 |
| Palizada River | 4.556 | 4.556 | 3.168 | 2 |
| Terminos Lagoon | 3.000 | 3.000 | 2.495 | 1 |
| Sea | 4.556 | 4.556 | 3.225 | 0 |
| Tzendales | Lacantun River | Chajul | Benemerito | Emiliano Zapata | San Pedro River | Chacamax | Jonuta | Canitzan | Pom Lagoon | Palizada River | Terminos Lagoon | Sea | |
| Tzendales | 0 | ||||||||||||
| Lacantun River | 0.015 | 0 | |||||||||||
| Chajul | 0.117 | 0.054 | 0 | ||||||||||
| Benemerito | 0.003 | 0.034 | 0.090 | 0 | |||||||||
| Emiliano Zapata | 0.089 | 0.054 | −0.013 | 0.067 | 0 | ||||||||
| San Pedro River | 0.020 | 0.025 | 0.088 | 0.085 * | 0.081 * | 0 | |||||||
| Chacamax | 0.093 | 0.065 | 0.063 | 0.104 * | 0.064 | 0.043 | 0 | ||||||
| Jonuta | 0.043 | 0.014 | 0.090 | 0.132 | 0.107 | −0.009 | 0.04 | 0 | |||||
| Canitzan | 0.046 | 0.027 | 0.069 | 0.078 | 0.082 * | −0.007 | 0.017 | −0.004 | 0 | ||||
| Pom Lagoon | 0.113 | 0.082 | 0.019 | 0.127 | −0.066 | 0.111 | 0.112 | 0.121 | 0.097 | 0 | |||
| Palizada River | 0.041 | 0.0002 | 0.015 | 0.091 | −0.004 | −0.002 | 0.037 | −0.003 | −0.001 | 0.011 | 0 | ||
| Terminos Lagoon | 0.004 | 0.025 | 0.101 * | −0.034 | 0.064 | 0.088 * | 0.115 | 0.084 | 0.094 * | 0.104 * | 0.068 * | 0 | |
| Sea | 0.051 | 0.038 | 0.106 | −0.02 | 0.102 | 0.125 | 0.103 | 0.141 | 0.114 * | 0.148 * | 0.118 * | −0.038 | 0 |
| From k = 1 | From k = 2 | From k = 3 | Ne | |
|---|---|---|---|---|
| DAPC | ||||
| To K = 1 | (m = 0.9745) 86.9254 | (m = 0.042) 3.7464 | (m = 0.0244) 2.17648 | 89.2 |
| To K = 2 | (m = 0.009) 0.0531 | (m = 0.9159) 5.40381 | (m = 0.0254) 0.14986 | 5.9 |
| To K = 3 | (m = 0.0164) 0.19516 | (m = 0.042) 0.4998 | (m = 0.9501) 11.30619 | 11.9 |
| STRUCTURE K = 3 | ||||
| To K = 1 | (m = 0.95581) 94.434028 | (m = 0.07456) 7.366528 | (m = 0.04342) 4.289896 | 98.8 |
| To K = 2 | (m = 0.00872) 0.142136 | (m = 0.88829) 14.479127 | (m = 0.02753) 0.448739 | 16.3 |
| To K = 3 | (m = 0.03545) 0.57429 | (m = 0.03719) 0.602478 | (m = 0.92904) 15.050448 | 16.2 |
| STRUCTURE K = 2 | ||||
| To K = 1 | (m = 0.98839) 23.128326 | (m = 0.0834) 1.95156 | N/A | 23.4 |
| To K = 2 | (m = 0.01161) 0.189243 | (m = 0.9166) 14.94058 | N/A | 16.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terán-Martínez, J.; Rodiles-Hernández, R.; Garduño-Sánchez, M.A.A.; Ornelas-García, C.P. Molecular Characterization of the Common Snook, Centropomus undecimalis (Bloch, 1792) in the Usumacinta Basin. Diversity 2021, 13, 347. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080347
Terán-Martínez J, Rodiles-Hernández R, Garduño-Sánchez MAA, Ornelas-García CP. Molecular Characterization of the Common Snook, Centropomus undecimalis (Bloch, 1792) in the Usumacinta Basin. Diversity. 2021; 13(8):347. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080347
Chicago/Turabian StyleTerán-Martínez, Jazmín, Rocío Rodiles-Hernández, Marco A. A. Garduño-Sánchez, and Claudia Patricia Ornelas-García. 2021. "Molecular Characterization of the Common Snook, Centropomus undecimalis (Bloch, 1792) in the Usumacinta Basin" Diversity 13, no. 8: 347. https://0-doi-org.brum.beds.ac.uk/10.3390/d13080347
{kind=link}