
Characterization and Phylogenetic Analyses of the Complete Mitochondrial Genome of Sugarcane (Saccharum spp. Hybrids) Line A1

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sampling, Mitochondria DNA Sequencing and Genome Assembly
2.2. Genome Annotation and Sequence Analyses
2.3. Repetitive Elements Analysis
2.4. RNA Editing Analyses
2.5. Phylogenetic Analysis
3. Results
3.1. Characteristics of the Mitogenome of Sugarcane Line A1
3.2. Codon Usage Bias
3.3. Repetitive Element and Simple Sequence Repeat (SSR) Analysis
3.4. The Prediction of RNA Editing
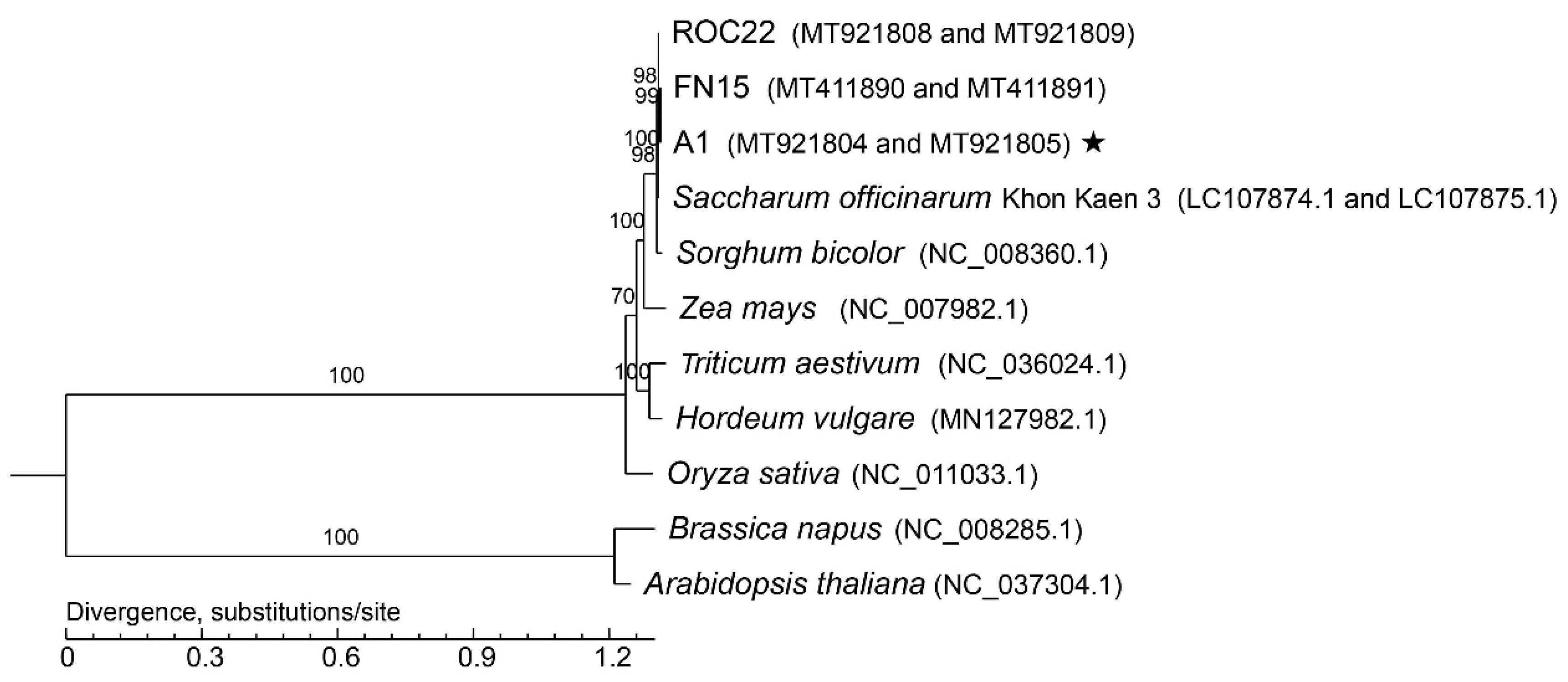
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Shtolz, N.; Mishmar, D. The mitochondrial genome–on selective constraints and signatures at the organism, cell, and single mitochondrion levels. Front. Ecol. 2019, 7, 342. [Google Scholar] [CrossRef] [Green Version]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.C. The mitochondrial compartment. J. Exp. Bot. 2007, 57, 1225–1243. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria—Past, present and future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef] [PubMed]
- Bergthorsson, U.; Richardson, A.O.; Young, G.J.; Goertzen, L.R.; Palmer, J.D. Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm Amborella. Proc. Natl. Acad. Sci. USA 2004, 101, 17747–17752. [Google Scholar] [CrossRef] [Green Version]
- Oda, K.; Katsuyuki, Y.; Ohta, E.; Nakamura, Y.; Takemura, M.; Nozato, N.; Akashi, K.; Kanegae, T.; Ogura, Y.; Kohchi, T.; et al. Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA. A primitive form of plant mitochondrial genome. J. Mol. Biol. 1992, 223, 1–7. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.T.; Foster, P.G.; Littlewood, D.T. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene 2014, 533, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kivisild, T. Maternal ancestry and population history from whole mitochondrial genomes. Investig. Genet. 2015, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; He, L.; Gao, S.; Su, Y.; Xu, L. Comparative analysis of two sugarcane ancestors Saccharum officinarum and S. spontaneum based on complete chloroplast genomesequences and photosynthetic ability in cold stress. Int. J. Mol. Sci. 2019, 20, 3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisano, H.; Tsujimura, M.; Yoshida, H.; Terachi, T.; Sato, K. Mitochondrial genome sequences from wild and cultivated barley (Hordeum vulgare). BMC Genom. 2016, 17, 824. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Liu, X.; Gao, S.; Guo, J.; Su, Y.; Ling, H.; Wang, C.; Li, Z.; Xu, L.; Que, Y. Foreign cry1Ac gene integration and endogenous borer stress-related genes synergistically improve insect resistance in sugarcane. BMC Plant Biol. 2018, 18, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hua, X.; Zhong, W.; Yuan, Y.; Wang, Y.; Wang, Z.; Ming, R.; Zhang, J. Genome-wide identification and expression profile analysis of WRKY family genes in the autopolyploid Saccharum spontaneum. Plant Cell Physiol. 2019, 3, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, Z.; Liu, Y.; Li, Z.; Zhou, D. The complete mitochondrial genome of sugarcane (Saccharum spp.) variety FN15. Mitochondrial DNA B Resour. 2020, 5, 2163–2165. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Z.; Lu, G.; Zeng, R.; Que, Y. Sugarcane ratooning ability:research satus, shortcomings, and prospects. Biology 2021, 10, 1052. [Google Scholar] [CrossRef]
- Irvine, J. Saccharum species as horticultural classes. Theor. Appl. Genet. 1999, 98, 186–194. [Google Scholar] [CrossRef]
- Bremer, G. Problems in breeding and cytology of sugar cane. Euphytica 1961, 10, 59–78. [Google Scholar] [CrossRef]
- Sobhakumari, V.P. New determinations of somatic chromosome number in cultivated and wild species of Saccharum. Caryologia 2013, 66, 268–274. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Q.; Li, L.; Tang, H.; Zhang, Q.; Chen, Y.; Arrow, J.; Zhang, X.; Wang, A.; Miao, C.; et al. Recent polyploidization events in three saccharum founding species. Plant Biotechnol. J. 2018, 17, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Hont, A. Unraveling the genome structure of polyploids using FISH and GISH; examples of sugarcane and banana. Cytogenet Genome Res. 2005, 109, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Garsmeur, O.; Droc, G.; Antonise, R.; Grimwood, J.; Potier, B.; Aitken, K.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C.; et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Yin, Z.; Liu, X.; Li, Z.; Yan, M.; Que, Y.; Xu, L. The complete mitochondrial genome sequence and phylogenetic analysis of sugarcane (Saccharum spp.) cultivar ROC22. Mitochondrial DNA Part B 2020, 5, 1915–1916. [Google Scholar] [CrossRef] [Green Version]
- Shearman, J.R.; Sonthirod, C.; Naktang, C.; Pootakham, W.; Yoocha, T.; Sangsrakru, D.; Jomchai, N.; Tragoonrung, S.; Tangphatsornruang, S. The two chromosomes of the mitochondrial genome of a sugarcane cultivar: Assembly and recombination analysis using long PacBio reads. Sci. Rep. 2016, 6, 31533. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Guan, R.; Chang, S.; Du, T.; Zhang, H.; Xing, H. Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS ONE 2011, 6, e17662. [Google Scholar] [CrossRef] [Green Version]
- Borgström, E.; Lundin, S.; Lundeberg, J. Large scale library generation for high throughput sequencing. PLoS ONE 2011, 6, e19119. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrane, M.; UniProt Consortium. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, bar009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [Green Version]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yu, J.; Yu, X.; Zhang, D.; Chang, H.; Li, W.; Song, H.; Cui, Z.; Wang, P.; Luo, Y.; et al. Structural variation of mitochondrial genomes sheds light on evolutionary history of soybeans. Plant J. 2021, 108, 1456–1472. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, M.; Kaneko, T.; Sakamoto, T.; Kimura, S.; Shigyo, M.; Yamagishi, H.; Terachi, T. Multichromosomal structure of the onion mitochondrial genome and a transcript analysis. Mitochondrion 2019, 46, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Gao, C.; Liu, J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genom. 2022, 23, 147. [Google Scholar] [CrossRef]
- Shidhi, P.R.; Biju, V.C.; Anu, S.; Vipin, C.L.; Deelip, K.R.; Achuthsankar, S.N. genome characterization, comparison and phylogenetic analysis of complete mitochondrial genome of evolvulus alsinoides reveals highly rearranged gene order in Solanales. Life 2021, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhu, A.; Fan, W.; Mower, J.P. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 2016, 213, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Mileshina, D.; Koulintchenko, M.; Konstantinov, Y.; Dietrich, A. Transfection of plant mitochondria and in organello gene integration. Nucleic Acids Res. 2011, 39, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; He, X.; Priyadarshani, S.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, M.S.; Omelchenko, D.O.; Usatov, A.V.; Gavrilova, V.A. The insights into mitochondrial genomes of sunflowers. Plants 2021, 10, 1774. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed Res. Int. 2019, 2019, 5040598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Wang, D.X.; Huang, Y.Q.; Chen, H.; Du, H.; Tu, J.M. Detection and analysis of C-to-U RNA Editing in rice mitochondria-encoded ORFs. Plants 2020, 9, 1277. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cuthbert, J.M.; Taylor, D.R.; Sloan, D.B. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef] [Green Version]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | SSR Type | SSR | Start | End | Location |
|---|---|---|---|---|---|
| A1-chr1 | p1 | (T)10 | 21,434 | 21,443 | IGS |
| A1-chr1 | p5 | (ATAGA)12 | 28,425 | 28,484 | IGS |
| A1-chr1 | p1 | (T)10 | 42,775 | 42,784 | IGS |
| A1-chr1 | p3 | (CTA)5 | 65,115 | 65,129 | IGS |
| A1-chr1 | p1 | (T)12 | 65,595 | 65,606 | IGS |
| A1-chr1 | p1 | (T)10 | 74,767 | 74,776 | nad1 |
| A1-chr1 | p3 | (ATA)5 | 76,342 | 76,356 | IGS |
| A1-chr1 | p1 | (T)14 | 138,736 | 138,749 | IGS |
| A1-chr1 | c | (T)11acttattaaattctctgtcttgctaaacacaaatccttcttttcttgtatagacg(A)11 | 145,252 | 145,328 | IGS |
| A1-chr1 | p1 | (A)11 | 153,201 | 153,211 | IGS |
| A1-chr1 | p1 | (T)10 | 166,287 | 166,296 | IGS |
| A1-chr1 | p1 | (A)13 | 178,747 | 178,759 | IGS |
| A1-chr1 | p1 | (T)10 | 184,327 | 184,336 | IGS |
| A1-chr1 | p1 | (T)10 | 208,192 | 208,201 | IGS |
| A1-chr1 | p2 | (GA)6 | 220,084 | 220,095 | IGS |
| A1-chr1 | p1 | (A)10 | 221,396 | 221,405 | IGS |
| A1-chr1 | p1 | (A)11 | 245,842 | 245,852 | IGS |
| A1-chr1 | p1 | (T)13 | 246,511 | 246,523 | rps1 |
| A1-chr1 | p1 | (A)10 | 256,149 | 256,158 | IGS |
| A1-chr1 | p1 | (A)12 | 262,049 | 262,060 | IGS |
| A1-chr1 | p1 | (T)10 | 280,624 | 280,633 | IGS |
| A1-chr2 | p1 | (G)10 | 12,943 | 12,952 | IGS |
| A1-chr2 | p1 | (T)10 | 26,230 | 26,239 | IGS |
| A1-chr2 | p1 | (T)10 | 30,036 | 30,045 | IGS |
| A1-chr2 | p1 | (T)10 | 34,409 | 34,418 | IGS |
| A1-chr2 | p1 | (A)10 | 37,040 | 37,049 | rps3 |
| A1-chr2 | p1 | (T)11 | 61,305 | 61,315 | IGS |
| A1-chr2 | p1 | (A)10 | 61,669 | 61,678 | IGS |
| A1-chr2 | p5 | (TAATA)14 | 67,753 | 67,822 | IGS |
| A1-chr2 | p1 | (T)12 | 80,494 | 80,505 | IGS |
| A1-chr2 | c | (T)10ctctccta(T)10 | 81,051 | 81,078 | IGS |
| A1-chr2 | c | (T)10agttcgcactgctctttctctctaaattgcatcaaagaaaat(AG)6 | 88,702 | 88,765 | IGS |
| A1-chr2 | p5 | (ATAGA)20 | 90,935 | 91,034 | IGS |
| A1-chr2 | p2 | (AT)7 | 105,454 | 105,467 | IGS |
| A1-chr2 | p5 | (GTATA)9 | 108,891 | 108,935 | IGS |
| A1-chr2 | p1 | (T)10 | 109,400 | 109,409 | IGS |
| A1-chr2 | p1 | (A)10 | 122,745 | 122,754 | IGS |
| A1-chr2 | p1 | (T)10 | 140,466 | 140,475 | IGS |
| Type | RNA-Editing | Number | Percentage |
|---|---|---|---|
| hydrophobic | CCA (P) = > CTA (L) | 38 | 30.85% |
| CCG (P) = > CTG (L) | 19 | ||
| CCT (P) = > CTT (L) | 24 | ||
| CCT (P) = > TTT (F) | 6 | ||
| CCC (P) = > TTC (F) | 8 | ||
| GCC (A) = > GTC (V) | 3 | ||
| GCG (A) = > GTG (V) | 2 | ||
| GCT (A) = > GTT (V) | 3 | ||
| GCA (A) = > GTA (V) | 2 | ||
| CTT (L) = > TTT (F) | 22 | ||
| CTC (L) = > TTC (F) | 10 | ||
| hydrophilic | CAT (H) = > TAT (Y) | 16 | 11.49% |
| CAC (H) = > TAC (Y) | 5 | ||
| CGT (R) = > TGT (C) | 25 | ||
| CGC (R) = > TGC (C) | 5 | ||
| hydrophobic-hydrophilic | CCG (P) = > TCG (S) | 9 | 12.16% |
| CCT (P) = > TCT (S) | 12 | ||
| CCA (P) = > TCA (S) | 8 | ||
| CCC (P) = > CTC (L) | 10 | ||
| CCC (P) = > TCC (S) | 15 | ||
| hydrophilic-hydrophobic | CGG (R) = > TGG (W) | 28 | 45.05% |
| TCC (S) = > TTC (F) | 31 | ||
| TCT (S) = > TTT (F) | 32 | ||
| TCA (S) = > TTA (L) | 51 | ||
| TCG (S) = > TTG (L) | 38 | ||
| ACC (T) = > ATC (I) | 2 | ||
| ACT (T) = > ATT (I) | 7 | ||
| ACA (T) = > ATA (I) | 7 | ||
| ACG (T) = > ATG (M) | 4 | ||
| hydrophilic-stop | CGA (R) = > TGA (X) | 1 | 0.45% |
| CAG (Q) = > TAG (X) | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, D.; Liu, Y.; Yao, J.; Yin, Z.; Wang, X.; Xu, L.; Que, Y.; Mo, P.; Liu, X. Characterization and Phylogenetic Analyses of the Complete Mitochondrial Genome of Sugarcane (Saccharum spp. Hybrids) Line A1. Diversity 2022, 14, 333. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050333
Zhou D, Liu Y, Yao J, Yin Z, Wang X, Xu L, Que Y, Mo P, Liu X. Characterization and Phylogenetic Analyses of the Complete Mitochondrial Genome of Sugarcane (Saccharum spp. Hybrids) Line A1. Diversity. 2022; 14(5):333. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050333
Chicago/Turabian StyleZhou, Dinggang, Ying Liu, Jingzuo Yao, Ze Yin, Xinwen Wang, Liping Xu, Youxiong Que, Ping Mo, and Xiaolan Liu. 2022. "Characterization and Phylogenetic Analyses of the Complete Mitochondrial Genome of Sugarcane (Saccharum spp. Hybrids) Line A1" Diversity 14, no. 5: 333. https://0-doi-org.brum.beds.ac.uk/10.3390/d14050333