
Rolling Circle Enhanced Detection of Specific Restriction Endonuclease Activities in Crude Cell Extracts

 , ,
, ,  and
and 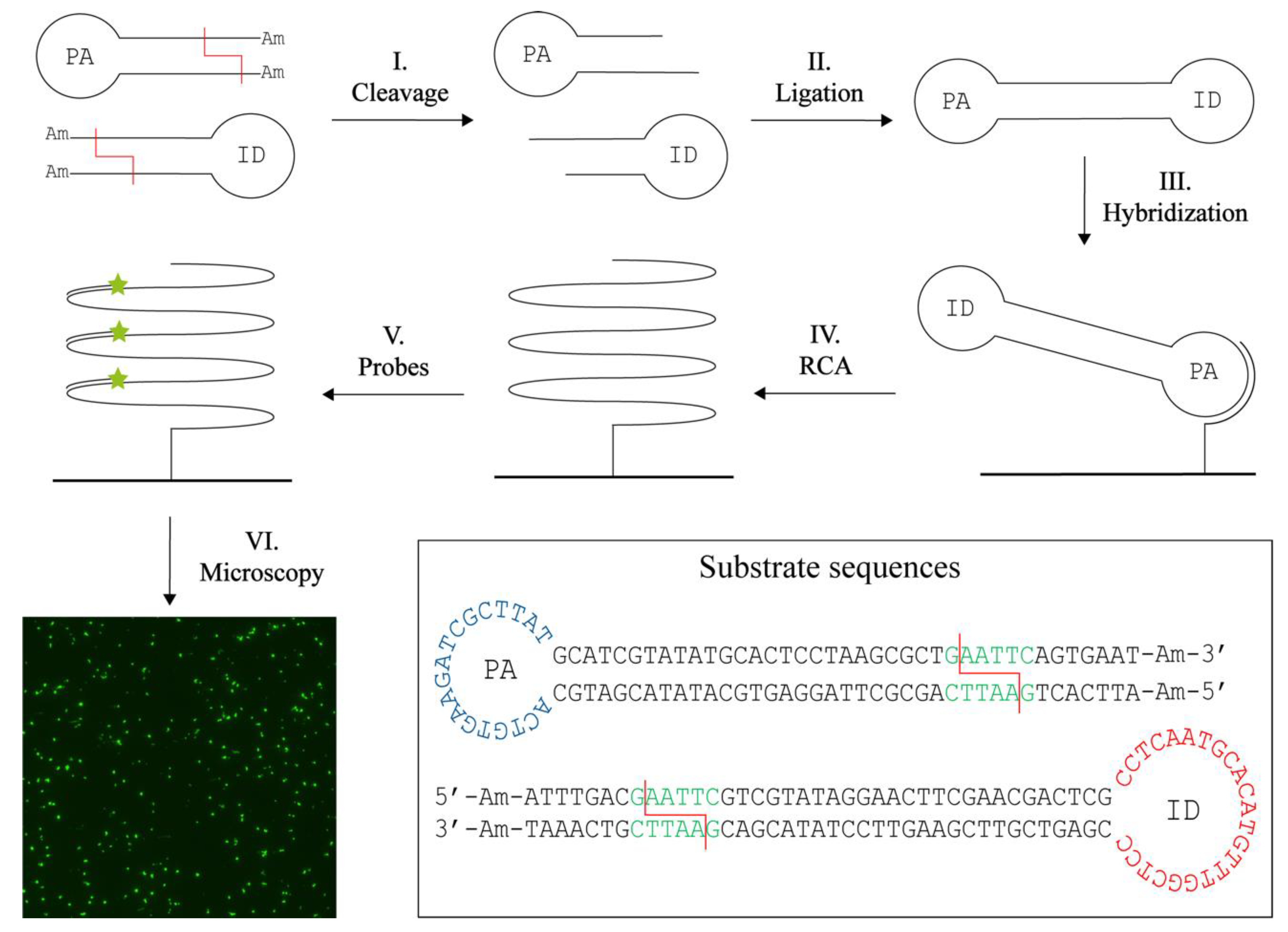{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. DNA Substrates and Oligonucleotides
- 5′-amine REEAD primer: 5′-/AmC6/CCAACCAACCAACCAAATAAGCGATCTTC
- ID Detection probe: 5′-/FAM/CCTCAATGCACATGTTTGGCTCC-3′
- T4 EcoRI PA substrate: 5′-/AmC6/ATTCACTgaattcAGCGCTTAGGAGTGCATATACGATGCACTGTGAAGATCGCTTATGCATCGTATATGCACTCCTAAGCGCT
- T4 EcoRI ID substrate: 5′-/AmC6/ATTTGACgaattcGTCGTATAGGAACTTCGAACGACTCGCCTCAATGCACATGTGGCTCCCGAGTCGTTCGAAGTTCCTATACGACgaattcGTCAAAT/AmC6/-3′
- T4 XhoI PA substrate: 5′-/AmC6/ATTCACTctcgagAGCGCTTAGGAGTGCATATACGATGCACTGTGAAGATCGCTTATGCATCGTATATGCACTCCTAAGCGCT
- T4 XhoI ID substrate: 5′-/AmC6/ATTTGACctcgagGTCGTATAGGAACTTCGAACGACTCGCCTCAATGCACATGTTTGGCTCC-CGAGTCGTTCGAAGTTCCTATAC
- Cre EcoRI PA substrate: 5′-/AmC6/GCGACgaattcAATTATACGAAGTTATTCGCA
- Cre XhoI PA substrate: 5′-/AmC6/GCGACctcgagAATTATACGAAGTTATTCGCA
- TACTGTGAAGATCGCTTATGAATAACTTCGTATAATTctcgagGTCGC/AmC6/-3′
2.3. Purification of T4 DNA Ligase
2.4. Purification of Cre Recombinase
2.5. Preparation of Bacterial Lysates
2.6. Preparation of Functionalized Glass Slide
2.7. T4 DNA Ligase Assisted RE Detection
2.8. Cre Assisted RE Detection
2.9. Statistics
3. Results and Discussion
3.1. T4 DNA Ligase Assisted Restriction Endonuclease Detection
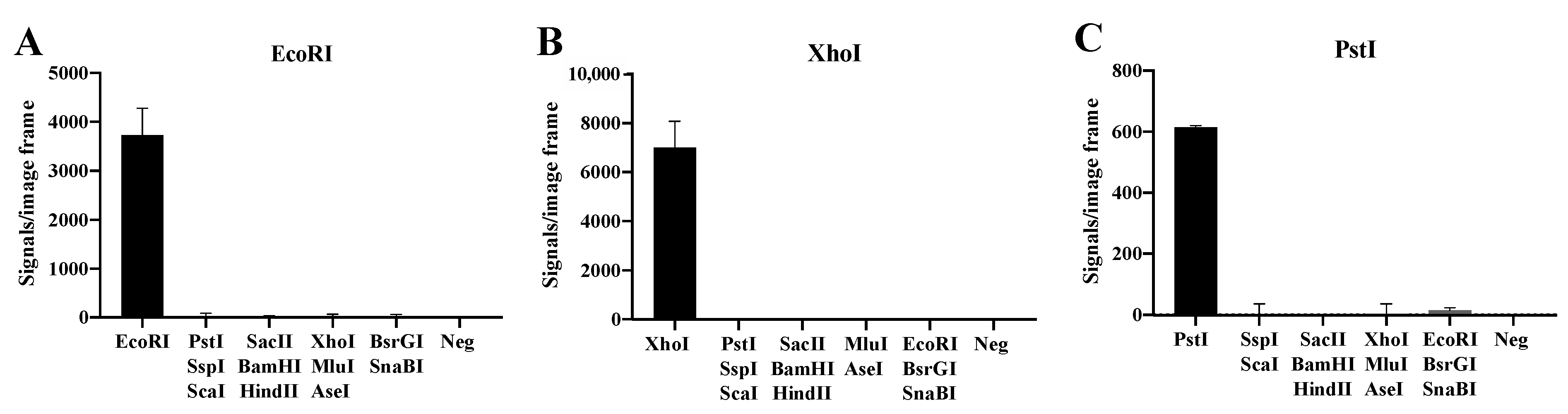
3.2. Specific Detection of Purified RE Activities
3.3. Detection of REs in Cell Extracts
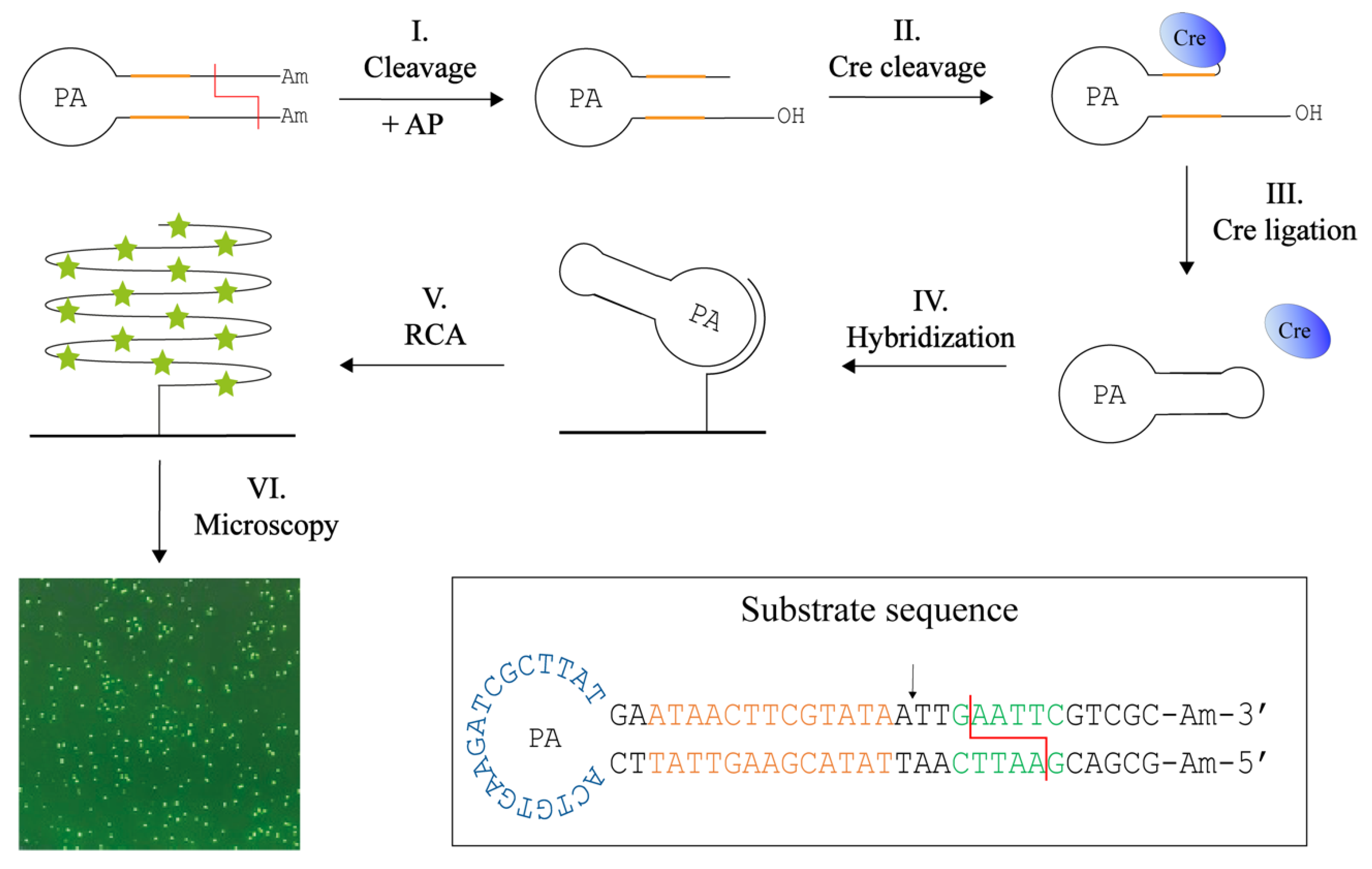
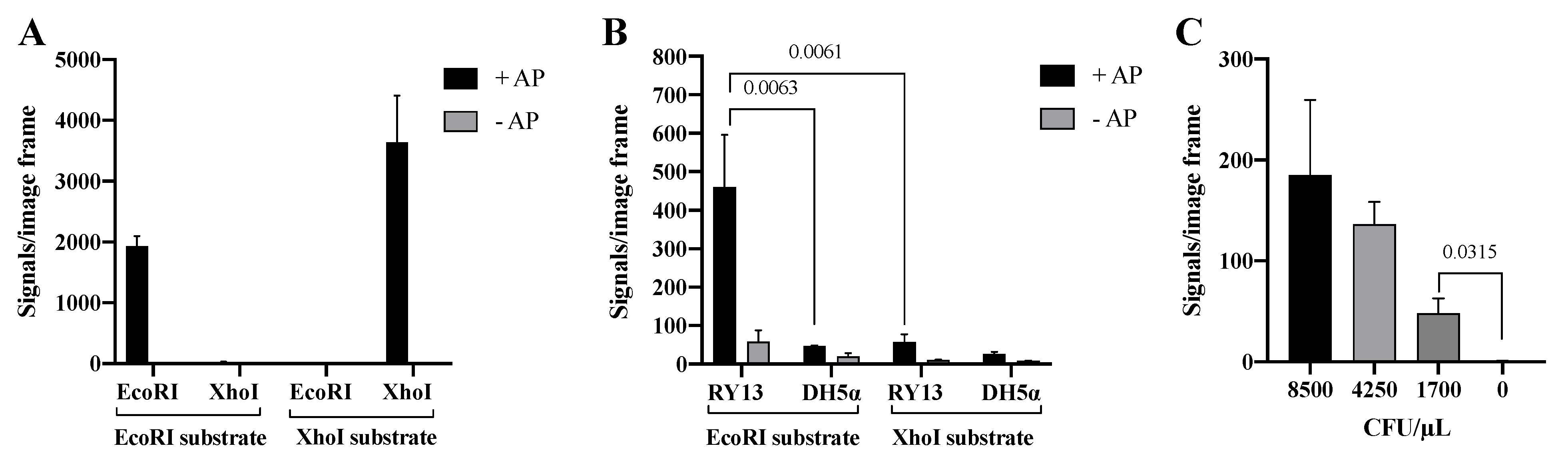
3.4. Specific End-Joining by Cre Recombinase
3.5. Detection of REs with Cre-Assisted RE Detection
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberts, R.J.; Murray, K. Restriction Endonuclease. Crit. Rev. Biotechnol. 1976, 4, 123–164. [Google Scholar] [CrossRef]
- Kessler, C.; Neumaier, P.S.; Wolf, W. Recognition sequences of restriction endonucleases and methylases—A review. Gene 1985, 33, 1–102. [Google Scholar] [CrossRef]
- McClelland, M. The effect of sequence specific DNA methylation on restriction endonuclease cleavage. Nucleic. Acids Res. 1981, 9, 5859–5866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenen, W.A.M.; Dryden, D.T.F.; Raleigh, E.A.; Wilson, G.G.; Murrayy, N.E. Highlights of the DNA cutters: A short history of the restriction enzymes. Nucleic Acids Res. 2014, 42, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingoud, A.; Jeltsch, A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001, 29, 3705–3727. [Google Scholar] [CrossRef]
- Pingoud, A.; Fuxreiter, M.; Pingoud, V.; Wende, W. Cellular and Molecular Life Sciences Type II restriction endonucleases: Structure and mechanism. Cell Mol. Life Sci. 2005, 62, 685–707. [Google Scholar] [CrossRef]
- Di Felice, F.; Micheli, G.; Camilloni, G. Restriction enzymes and their use in molecular biology: An overview. J. Biosci. 2019, 44, 1–8. [Google Scholar] [CrossRef]
- Roberts, R.J. How restriction enzymes became the workhorses of molecular biology. Proc. Natl. Acad. Sci. USA 2005, 102, 5905–5908. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D.; Biolabs, N.E. REBASE––A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, 2014–2015. [Google Scholar] [CrossRef] [PubMed]
- Southern, E. Gel electrophoresis of restriction fragments. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1979; Volume 68, pp. 152–176. ISBN 0076-6879. [Google Scholar]
- Jeltsch, A.; Fritz, A.; Alves, J.; Wolfes, H.; Pingoud, A. A fast and accurate enzyme-linked immunosorbent assay for the determination of the DNA cleavage activity of restriction endonucleases. Anal. Biochem. 1993, 213, 234–240. [Google Scholar] [CrossRef]
- Jeltsch, A.; Pingoud, A.M. Methods for determining activity and specificity of DNA binding and DNA cleavage by class II restriction endonucleases. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2001; pp. 287–308. ISBN 978-1-59259-233-3. [Google Scholar]
- Ghosh, S.S.; Eis, P.S.; Blumeyer, K.; Fearon, K.; Millar, D.P. Real time kinetics of restriction endonuclease cleavage monitored by fluorescence resonance energy transfer. Nucleic Acids Res. 1994, 22, 3155–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenschmidt, K.; Lanio, T.; Jeltsch, A.; Pingoud, A. A fluorimetric assay for on-line detection of DNA cleavage by restriction endonucleases. J. Biotechnol. 2002, 96, 185–191. [Google Scholar] [CrossRef]
- Ma, C.; Tang, Z.; Wang, K.; Tan, W.; Yang, X.; Li, W.; Li, Z.; Lv, X. Real-time monitoring of restriction endonuclease activity using molecular beacon. Anal. Biochem. 2007, 363, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Stougaard, M.; Lohmann, J.S.; Mancino, A.; Andersen, F.F.; Koch, J.; Knudsen, B.R. Single-Molecule Detection of Human Topoisomerase I Cleavage-Ligation Activity. ACS Nano 2009, 3, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Hede, M.S.; Fjelstrup, S.; Lotsch, F.; Zoleko, R.M.; Klicpera, A.; Groger, M.; Mischlinger, J.; Endame, L.; Veletzky, L.; Neher, R.; et al. Detection of the Malaria Causing Plasmodium parasite in Saliva from Infected Patients Using Topoisomerase I Activity as a Biomarker. Sci. Rep. 2018, 8, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juul, S.; Nielsen, C.J.F.; Labouriau, R.; Roy, A.; Tesauro, C.; Jensen, P.W.; Harmsen, C.; Kristoffersen, E.L.; Chiu, Y.-L.; Frohlich, R.; et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing Plasmodium parasites based on enzyme activity measurement. ACS Nano 2012, 6, 10676–10683. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, J.; Thomsen, J.; Selnihhin, D.; Hede, M.S.; Kirsebom, F.C.M.; Franch, O.; Fjelstrup, S.; Stougaard, M.; Ho, Y.-P.; et al. Novel DNA sensor system for highly sensitive and quantitative retrovirus detection using virus encoded integrase as a biomarker. Nanoscale 2017, 9, 440–448. [Google Scholar] [CrossRef]
- Franch, O.; Han, X.; Marcussen, L.B.; Givskov, A.; Andersen, M.B.; Godbole, A.A.; Harmsen, C.; Nørskov-Lauritsen, N.; Thomsen, J.; Pedersen, F.S.; et al. A new DNA sensor system for specific and quantitative detection of mycobacteria. Nanoscale 2019, 11, 587–597. [Google Scholar] [CrossRef]
- Chen, P.; den Bakker, H.C.; Korlach, J.; Kong, N.; Storey, D.B.; Paxinos, E.E.; Ashby, M.; Clark, T.; Luong, K.; Wiedmann, M.; et al. Comparative Genomics Reveals the Diversity of Restriction-Modification Systems and DNA Methylation Sites in Listeria monocytogenes. Appl. Environ. Microbiol. 2017, 83, e02091–e02116. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Morgan, R.D.; Roberts, R.J.; Blaser, M.J. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc. Natl. Acad. Sci. USA 2000, 97, 9671–9676. [Google Scholar] [CrossRef]
- Modrich, P.; Zabel, D. EcoRI endonuclease. Physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 1976, 251, 5866–5874. [Google Scholar] [CrossRef]
- Kuhn, H.; Frank-Kamenetskii, M.D. Template-independent ligation of single-stranded DNA by T4 DNA ligase. FEBS J. 2005, 272, 5991–6000. [Google Scholar] [CrossRef] [PubMed]
- Western, L.M.; Rose, S.J. A novel DNA joining activity catalyzed by T4 DNA ligase. Nucleic Acids Res. 1991, 19, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Duyne, G.D. Cre Recombinase. Microbiol. Spectr. 2015, 3, MDNA3-0014-2014. [Google Scholar] [CrossRef]
- Hoess, R.H.; Abremski, K. Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J. Mol. Biol. 1985, 181, 351–362. [Google Scholar] [CrossRef]
- Ma, C.-H.; Kachroo, A.H.; Macieszak, A.; Chen, T.-Y.; Guga, P.; Jayaram, M. Reactions of Cre with methylphosphonate DNA: Similarities and contrasts with Flp and vaccinia topoisomerase. PLoS ONE 2009, 4, e7248. [Google Scholar] [CrossRef] [Green Version]
- Andersen, F.F.; Stougaard, M.; Jorgensen, H.L.; Bendsen, S.; Juul, S.; Hald, K.; Andersen, A.H.; Koch, J.; Knudsen, B.R. Multiplexed detection of site specific recombinase and DNA topoisomerase activities at the single molecule level. ACS Nano 2009, 3, 4043–4054. [Google Scholar] [CrossRef]
- Keller, J.G.; Petersen, K.V.; Knudsen, B.R.; Tesauro, C. Simple and Fast DNA-Based Tool to Investigate Topoisomerase 1 Activity, a Biomarker for Drug Susceptibility in Colorectal Cancer. In Recent Understanding of Colorectal Cancer Treatment [Working Title]; IntechOpen: Rijeka, Croatia, 2022; Chapter 5. [Google Scholar] [CrossRef]
- Rice, P.A. Serine Resolvases. Microbiol. Spectr. 2015, 3, MDNA3-0045-2014. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen, K.V.; Tesauro, C.; Hede, M.S.; Pages, C.; Marcussen, L.B.; Keller, J.G.; Bugge, M.; Holm, K.; Bjergbæk, L.; Stougaard, M.; et al. Rolling Circle Enhanced Detection of Specific Restriction Endonuclease Activities in Crude Cell Extracts. Sensors 2022, 22, 7763. https://0-doi-org.brum.beds.ac.uk/10.3390/s22207763
Petersen KV, Tesauro C, Hede MS, Pages C, Marcussen LB, Keller JG, Bugge M, Holm K, Bjergbæk L, Stougaard M, et al. Rolling Circle Enhanced Detection of Specific Restriction Endonuclease Activities in Crude Cell Extracts. Sensors. 2022; 22(20):7763. https://0-doi-org.brum.beds.ac.uk/10.3390/s22207763
Chicago/Turabian StylePetersen, Kamilla Vandsø, Cinzia Tesauro, Marianne Smedegaard Hede, Camilla Pages, Lærke Bay Marcussen, Josephine Geertsen Keller, Magnus Bugge, Kasper Holm, Lotte Bjergbæk, Magnus Stougaard, and et al. 2022. "Rolling Circle Enhanced Detection of Specific Restriction Endonuclease Activities in Crude Cell Extracts" Sensors 22, no. 20: 7763. https://0-doi-org.brum.beds.ac.uk/10.3390/s22207763