
Measurement of Secondary Structure Changes in Poly-L-lysine and Lysozyme during Acoustically Levitated Single Droplet Drying Experiments by In Situ Raman Spectroscopy


Abstract
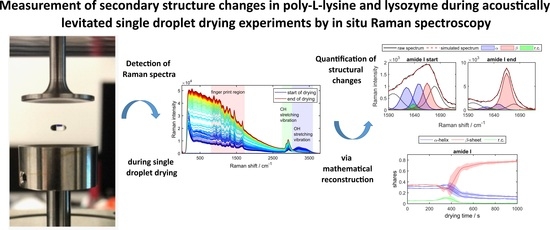
:
1. Introduction
2. Materials and Methods
2.1. Proteins and Sample Preparation
2.2. Experimental Setup and Measurement Procedure
3. Results and Discussion
3.1. Drying Protein Droplet Spectra
3.2. Secondary Structure Shares from Mathematical Reconstruction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secondary Structure | Amide I | Amide III | ||
|---|---|---|---|---|
| Poly-L-lysine | ||||
| This Work | Literature | This Work | Literature | |
| α-helix | 1625 | 1635 [50] | 1278 | 1276 [41] |
| 1649 | 1641 [56] | 1292 | 1305 [57] | |
| β-sheet | 1665 | 1665 [56] | 1241 | 1237 [56] |
| random coil | 1638 | 1641 [55] | 1257 | 1257 [47] |
| lysozyme | ||||
| α-helix | 1625 | 1634 [50] | 1278 | 1273 [57] |
| 1653 | 1655 [55] | 1295 | 1304 [57] | |
| β-sheet | 1667 | 1669 [58] | 1235 | 1229 [47] |
| random coil | 1635 | 1639 [55] | 1259 | 1250 [47] |
3.3. Temporal Changes of the Protein Structure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maa, Y.-F.; Prestrelski, S.J. Biopharmaceutical Powders Particle Formation and Formulation Considerations. Curr. Pharm. Biotechnol. 2000, 1, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Prestrelski, S.; Tedeschi, N.; Arakawa, T.; Carpenter, J. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 1993, 65, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Branchu, S.; Forbes, R.T.; York, P.; Petrén, S.; Nyqvist, H.; Camber, O. Hydroxypropyl-β-cyclodextrin inhibits spray-drying-induced inactivation of β-galactosidase. J. Pharm. Sci. 1999, 88, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Cal, K.; Sollohub, K. Spray Drying Technique. I: Hardware and Process Parameters. J. Pharm. Sci. 2010, 99, 575–586. [Google Scholar] [CrossRef]
- Perdana, J.; Fox, M.B.; Schutyser, M.A.I.; Boom, R.M. Single-Droplet Experimentation on Spray Drying: Evaporation of a Sessile Droplet. Chem. Eng. Technol. 2011, 34, 1151–1158. [Google Scholar] [CrossRef]
- Charlesworth, D.H.; Marshall, W.R. Evaporation from drops containing dissolved solids. AIChE J. 1960, 6, 9–23. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.M. Optical Levitation by Radiation Pressure. Appl. Phys. Lett. 1971, 19, 283–285. [Google Scholar] [CrossRef]
- Simon, M.D.; Geim, A.K. Diamagnetic levitation: Flying frogs and floating magnets (invited). J. Appl. Phys. 2000, 87, 6200–6204. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Wi, H.S.; Jo, W.; Cho, Y.C.; Lee, H.H.; Jeong, S.-Y.; Kim, Y.-I.; Lee, G.W. Multiple pathways of crystal nucleation in an extremely supersaturated aqueous potassium dihydrogen phosphate (KDP) solution droplet. Proc. Natl. Acad. Sci. USA 2016, 113, 13618–13623. [Google Scholar] [CrossRef] [Green Version]
- Hennet, L.; Cristiglio, V.; Kozaily, J.; Pozdnyakova, I.; Fischer, H.E.; Bytchkov, A.; Drewitt, J.W.E.; Leydier, M.; Thiaudière, D.; Grüner, S.; et al. Aerodynamic levitation and laser heating: Applications at synchrotron and neutron sources. Eur. Phys. J. Spec. Top. 2011, 196, 151. [Google Scholar] [CrossRef]
- Santesson, S.; Nilsson, S. Airborne chemistry: Acoustic levitation in chemical analysis. Anal. Bioanal. Chem. 2004, 378, 1704–1709. [Google Scholar] [CrossRef]
- Welter, E.; Neidhart, B. Acoustically levitated droplets—A new tool for micro and trace analysis. Fresenius J. Anal. Bioanal. Chem. 1997, 357, 345–350. [Google Scholar] [CrossRef]
- Bücks, K.; Müller, H. Über einige Beobachtungen an schwingenden Piezoquarzen und ihrem Schallfeld. Zeitschrift Physik 1933, 84, 75–86. [Google Scholar] [CrossRef]
- Tsujino, S.; Tomizaki, T. Ultrasonic acoustic levitation for fast frame rate X-ray protein crystallography at room temperature. Sci. Rep. 2016, 6, 25558. [Google Scholar] [CrossRef]
- Delißen, F.; Leiterer, J.; Bienert, R.; Emmerling, F.; Thünemann, A.F. Agglomeration of proteins in acoustically levitated droplets. Anal. Bioanal. Chem. 2008, 392, 161–165. [Google Scholar] [CrossRef]
- Leiterer, J.; Delißen, F.; Emmerling, F.; Thünemann, A.F.; Panne, U. Structure analysis using acoustically levitated droplets. Anal. Bioanal. Chem. 2008, 391, 1221–1228. [Google Scholar] [CrossRef]
- Omrane, A.; Santesson, S.; Aldén, M.; Nilsson, S. Laser techniques in acoustically levitated micro droplets. Lab Chip 2004, 4, 287–291. [Google Scholar] [CrossRef]
- Tuckermann, R.; Bauerecker, S.; Neidhart, B. Evaporation rates of alkanes and alkanols from acoustically levitated drops. Anal. Bioanal. Chem. 2001, 372, 122–127. [Google Scholar] [CrossRef]
- Kastner, O.; Brenn, G.; Rensink, D.; Tropea, C. The Acoustic Tube Levitator—A Novel Device for Determining the Drying Kinetics of Single Droplets. Chem. Eng. Technol. 2001, 24, 335–339. [Google Scholar] [CrossRef]
- Perlitz, J.; Broß, H.; Will, S. Measurement of Water Mole Fraction from Acoustically Levitated Pure Water and Protein Water Solution Droplets via Tunable Diode Laser Absorption Spectroscopy (TDLAS) at 1.37 µm. Appl. Sci. 2021, 11, 5036. [Google Scholar] [CrossRef]
- Brotton, S.J.; Kaiser, R.I. Novel high-temperature and pressure-compatible ultrasonic levitator apparatus coupled to Raman and Fourier transform infrared spectrometers. Rev. Sci. Instrum. 2013, 84, 055114. [Google Scholar] [CrossRef] [Green Version]
- Schenk, J.; Tröbs, L.; Emmerling, F.; Kneipp, J.; Panne, U.; Albrecht, M. Simultaneous UV/Vis spectroscopy and surface enhanced Raman scattering of nanoparticle formation and aggregation in levitated droplets. Anal. Methods 2012, 4, 1252–1258. [Google Scholar] [CrossRef]
- Quiño, J.; Hellwig, T.; Griesing, M.; Pauer, W.; Moritz, H.-U.; Will, S.; Braeuer, A. One-dimensional Raman spectroscopy and shadowgraphy for the analysis of the evaporation behavior of acetone/water drops. Int. J. Heat Mass Transf. 2015, 89, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Baer, S.; Esen, C.; Ostendorf, A. Phase equilibrium measurements of acoustically levitated squalane-CO2 mixtures by Raman spectroscopy. J. Raman Spectrosc. 2014, 45, 680–685. [Google Scholar] [CrossRef]
- Puskar, L.; Tuckermann, R.; Frosch, T.; Popp, J.; Ly, V.; McNaughton, D.; Wood, B.R. Raman acoustic levitation spectroscopy of red blood cells and Plasmodium falciparum trophozoites. Lab Chip 2007, 7, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.R.; Heraud, P.; Stojkovic, S.; Morrison, D.; Beardall, J.; McNaughton, D. A Portable Raman Acoustic Levitation Spectroscopic System for the Identification and Environmental Monitoring of Algal Cells. Anal. Chem. 2005, 77, 4955–4961. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.D.S.; Braeuer, A.; Arlabosse, P.; Ré, M.-I. In situ Raman composition profiling in drying droplets. Powder Technol. 2020, 373, 232–241. [Google Scholar] [CrossRef]
- Zölls, S.; Tantipolphan, R.; Wiggenhorn, M.; Winter, G.; Jiskoot, W.; Friess, W.; Hawe, A. Particles in Therapeutic Protein Formulations, Part 1: Overview of Analytical Methods. J. Pharm. Sci. 2012, 101, 914–935. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Williams, R.W. Protein secondary structure analysis using Raman amide I and amide III spectra. In Enzyme Structure; Hirs, C.H.W., Ed.; Academy Press: Orlando, FL, USA, 1986; pp. 311–331. ISBN 9780121820305. [Google Scholar]
- Wüthrich, K. Protein structure determination in solution by NMR spectroscopy. J. Biol. Chem. 1990, 265, 22059–22062. [Google Scholar] [CrossRef]
- Ilari, A.; Savino, C. Protein Structure Determination by X-Ray Crystallography. In Bioinformatics; Keith, J.M., Ed.; Humana Press LLC: Totowa, NJ, USA, 2008; Volume 452, pp. 63–87. [Google Scholar] [CrossRef]
- Lima, D.B.; Melchior, J.T.; Morris, J.; Barbosa, V.C.; Chamot-Rooke, J.; Fioramonte, M.; Souza, T.A.C.B.; Fischer, J.S.G.; Gozzo, F.C.; Carvalho, P.C.; et al. Characterization of homodimer interfaces with cross-linking mass spectrometry and isotopically labeled proteins. Nat. Protoc. 2018, 13, 431–458. [Google Scholar] [CrossRef] [Green Version]
- Pelton, J.T.; McLean, L.R. Spectroscopic Methods for Analysis of Protein Secondary Structure. Anal. Biochem. 2000, 277, 167–176. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Schrader, B. Infrared and Raman Spectroscopy: Methods and Applications; Wiley-VCH: Weinheim, Germany, 1995; ISBN 9783527615421. [Google Scholar]
- Casier, R.; Duhamel, J. Effect of Structure on Polypeptide Blobs: A Model Study Using Poly(l-lysine). Langmuir 2020, 36, 7980–7990. [Google Scholar] [CrossRef]
- Jackson, M.; Haris, P.I.; Chapman, D. Conformational transitions in poly(l-lysine): Studies using Fourier transform infrared spectroscopy. BBA Protein Struct. Mol. Enzym. 1989, 998, 75–79. [Google Scholar] [CrossRef]
- Mirtič, A.; Grdadolnik, J. The structure of poly-l-lysine in different solvents. Biophys. Chem. 2013, 175–176, 47–53. [Google Scholar] [CrossRef]
- Kambara, O.; Tamura, A.; Naito, A.; Tominaga, K. Structural changes of poly-l-lysine in solution and lyophilized form. Phys. Chem. Chem. Phys. 2008, 10, 5042–5044. [Google Scholar] [CrossRef]
- Ma, L.; Ahmed, Z.; Mikhonin, A.A.V.; Asher, S.A. UV Resonance Raman Measurements of Poly-l-Lysine’s Conformational Energy Landscapes: Dependence on Perchlorate Concentration and Temperature. J. Phys. Chem. B 2007, 111, 7675–7680. [Google Scholar] [CrossRef]
- Elkordy, A.; Forbes, R.T.; Barry, B.W. Stability of crystallised and spray-dried lysozyme. Int. J. Pharm. 2004, 278, 209–219. [Google Scholar] [CrossRef]
- Hedoux, A.; Ionov, R.; Willart, J.-F.; Lerbret, A.; Affouard, F.; Guinet, Y.; Descamps, M.; Prévost, D.; Paccou, L.; Danéde, F. Evidence of a two-stage thermal denaturation process in lysozyme: A Raman scattering and differential scanning calorimetry investigation. J. Chem. Phys. 2006, 124, 014703. [Google Scholar] [CrossRef]
- Dolui, S.; Mondal, A.; Roy, A.; Pal, U.; Das, S.; Saha, A.; Maiti, N.C. Order, Disorder, and Reorder State of Lysozyme: Aggregation Mechanism by Raman Spectroscopy. J. Phys. Chem. B 2019, 124, 50–60. [Google Scholar] [CrossRef]
- Maiti, N.C.; Apetri, M.M.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Raman Spectroscopic Characterization of Secondary Structure in Natively Unfolded Proteins: α-Synuclein. J. Am. Chem. Soc. 2004, 126, 2399–2408. [Google Scholar] [CrossRef]
- Alix, A.; Pedanou, G.; Berjot, M. Fast determination of the quantitative secondary structure of proteins by using some parameters of the Raman Amide I band. J. Mol. Struct. 1988, 174, 159–164. [Google Scholar] [CrossRef]
- Voicescu, M.; Ionescu, S.; Nistor, C.L. Spectroscopic study of 3-Hydroxyflavone-protein interaction in lipidic bi-layers immobilized on silver nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 170, 1–8. [Google Scholar] [CrossRef]
- Ngarize, S.; Herman, H.; Adams, A.; Howell, N. Comparison of Changes in the Secondary Structure of Unheated, Heated, and High-Pressure-Treated β-Lactoglobulin and Ovalbumin Proteins Using Fourier Transform Raman Spectroscopy and Self-Deconvolution. J. Agric. Food Chem. 2004, 52, 6470–6477. [Google Scholar] [CrossRef]
- Braeuer, P.A.; Bahr, L.A.; Röhricht, M.-L.; Schmidt, M.; Will, S. Spatially-resolved crystallinity determination of polymer welding seams by Raman-microscopy. Procedia CIRP 2020, 94, 796–801. [Google Scholar] [CrossRef]
- Movasaghi, Z.; Rehman, S.; Rehman, I.U. Raman Spectroscopy of Biological Tissues. Appl. Spectrosc. Rev. 2007, 42, 493–541. [Google Scholar] [CrossRef]
- Painter, P.C.; Koenig, J.L. The solution conformation of poly(L-lysine). A Raman and infrared spectroscopic study. Biopolymers 1976, 15, 229–240. [Google Scholar] [CrossRef]
- Abrarov, S.; Quine, B. Efficient algorithmic implementation of the Voigt/complex error function based on exponential series approximation. Appl. Math. Comput. 2011, 218, 1894–1902. [Google Scholar] [CrossRef]
- Abrarov, S.M.; Quine, B.M. On the Fourier expansion method for highly accurate computation of the Voigt/complex error function in a rapid algorithm. arXiv 2012, arXiv:1205.1768. [Google Scholar]
- Bauer, F.J.; Daun, K.J.; Huber, F.J.T.; Will, S. Can soot primary particle size distributions be determined using laser-induced incandescence? Appl. Phys. A 2019, 125, 109. [Google Scholar] [CrossRef]
- Sane, S.U.; Cramer, S.M.; Przybycien, T.M. A Holistic Approach to Protein Secondary Structure Characterization Using Amide I Band Raman Spectroscopy. Anal. Biochem. 1999, 269, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Carrier, D.; Pézolet, M. Raman spectroscopic study of the interaction of poly-L-lysine with dipalmitoylphosphatidylglycerol bilayers. Biophys. J. 1984, 46, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.M. Raman Spectroscopy for Monitoring Protein Structure in Muscle Food Systems. Crit. Rev. Food Sci. Nutr. 2008, 48, 512–523. [Google Scholar] [CrossRef]
- Ye, H.; Rahul; Kruger, U.; Wang, T.; Shi, S.; Norfleet, J.; De, S. Burn-Related Collagen Conformational Changes in Ex Vivo Porcine Skin Using Raman Spectroscopy. Sci. Rep. 2019, 9, 19138. [Google Scholar] [CrossRef] [Green Version]
- Czamara, K.; Petko, F.; Baranska, M.; Kaczor, A. Raman microscopy at the subcellular level: A study on early apoptosis in endothelial cells induced by Fas ligand and cycloheximide. Analyst 2016, 141, 1390–1397. [Google Scholar] [CrossRef]
- Qin, Z.; Buehler, M.J. Molecular Dynamics Simulation of theα-Helix toβ-Sheet Transition in Coiled Protein Filaments: Evidence for a Critical Filament Length Scale. Phys. Rev. Lett. 2010, 104, 198304. [Google Scholar] [CrossRef]
- Mauerer, A.; Lee, G. Changes in the amide I FT-IR bands of poly-l-lysine on spray-drying from α-helix, β-sheet or random coil conformations. Eur. J. Pharm. Biopharm. 2006, 62, 131–142. [Google Scholar] [CrossRef]
- Mattos, C. Protein–water interactions in a dynamic world. Trends Biochem. Sci. 2002, 27, 203–208. [Google Scholar] [CrossRef]
- Haque, M.A.; Adhikari, B. Drying and Denaturation of Proteins in Spray Drying Process. In Handbook of Industrial Drying; Mujumdar, A.S., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 971–983. ISBN 9780429169762. [Google Scholar]
- Martin, J.; Langer, T.; Boteva, R.; Schramel, A.; Horwich, A.L.; Hartl, F.-U. Chaperonin-mediated protein folding at the surface of groEL through a “molten globule”-like intermediate. Nature 1991, 352, 36–42. [Google Scholar] [CrossRef]
- Kocherbitov, V.; Latynis, J.; Misiūnas, A.; Barauskas, J.; Niaura, G. Hydration of Lysozyme Studied by Raman Spectroscopy. J. Phys. Chem. B 2013, 117, 4981–4992. [Google Scholar] [CrossRef]
- David, C.; Foley, S.; Enescu, M. Protein S–S bridge reduction: A Raman and computational study of lysozyme interaction with TCEP. Phys. Chem. Chem. Phys. 2009, 11, 2532–2542. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perlitz, J.F.A.; Gentner, L.; Braeuer, P.A.B.; Will, S. Measurement of Secondary Structure Changes in Poly-L-lysine and Lysozyme during Acoustically Levitated Single Droplet Drying Experiments by In Situ Raman Spectroscopy. Sensors 2022, 22, 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/s22031111
Perlitz JFA, Gentner L, Braeuer PAB, Will S. Measurement of Secondary Structure Changes in Poly-L-lysine and Lysozyme during Acoustically Levitated Single Droplet Drying Experiments by In Situ Raman Spectroscopy. Sensors. 2022; 22(3):1111. https://0-doi-org.brum.beds.ac.uk/10.3390/s22031111
Chicago/Turabian StylePerlitz, Julian F. A., Lukas Gentner, Phillipp A. B. Braeuer, and Stefan Will. 2022. "Measurement of Secondary Structure Changes in Poly-L-lysine and Lysozyme during Acoustically Levitated Single Droplet Drying Experiments by In Situ Raman Spectroscopy" Sensors 22, no. 3: 1111. https://0-doi-org.brum.beds.ac.uk/10.3390/s22031111