A Proline-Based Tectons and Supramolecular Synthons for Drug Design 2.0: A Case Study of ACEI

 , , ,
, , ,
Abstract
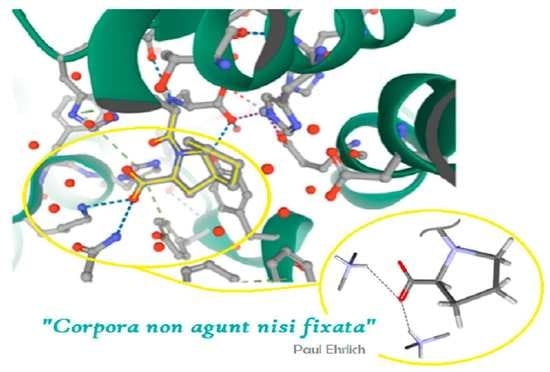
:
1. Introduction
1.1. Proline as Unique Amino Acid: Conformational Inclinations
1.2. Chirality of Proline
1.3. Zwiterion vs. Neutral Form of Proline
1.4. Proline Cis-Trans Isomerization
1.5. Further Insight into the Biological and Medicinal Relevance of Proline
1.6. Inhibitory Activity of Proline
1.7. ACE Inhibitors: Recent Updates
1.8. ACEI vs. Coronavirus Studies
1.9. Supramolecular Synthon Engineering for the Design of Drugs 2.0
2. Results and Discussion
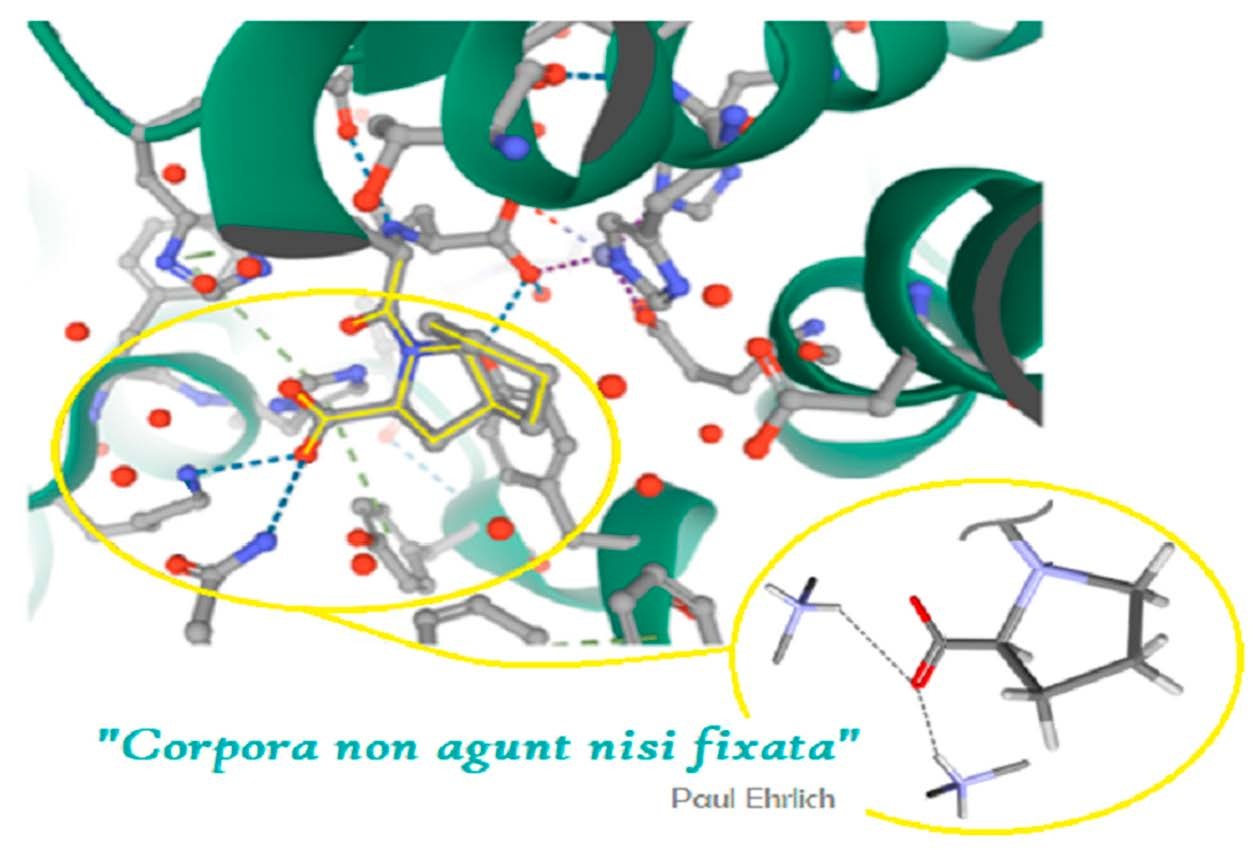
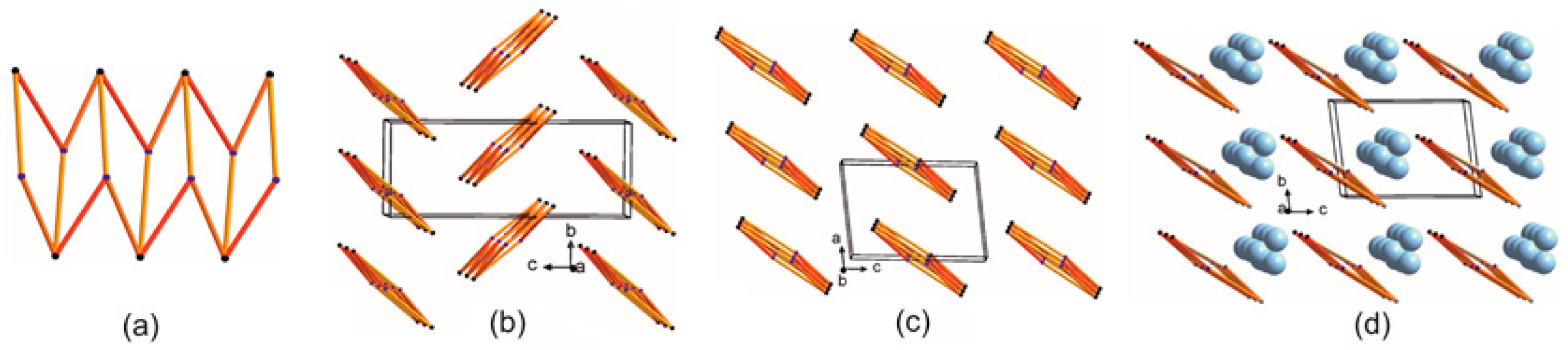
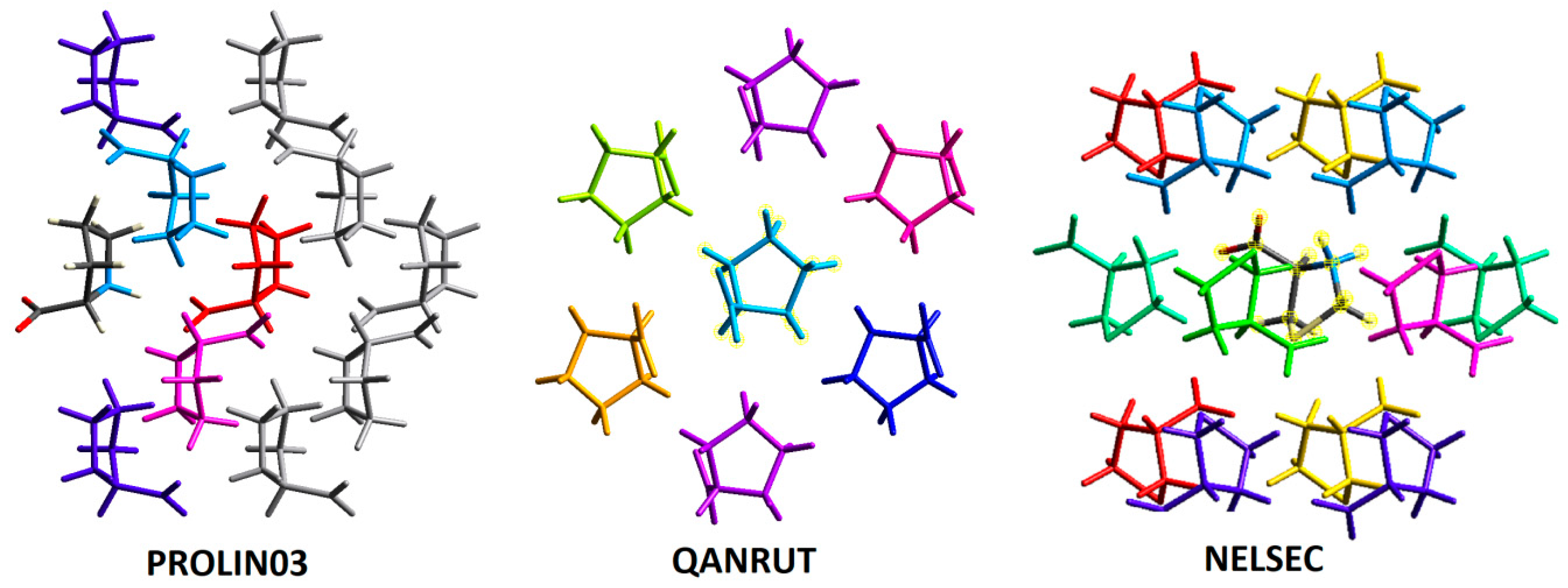
2.1. Molecular Focus on Proline Tectons: Database Story
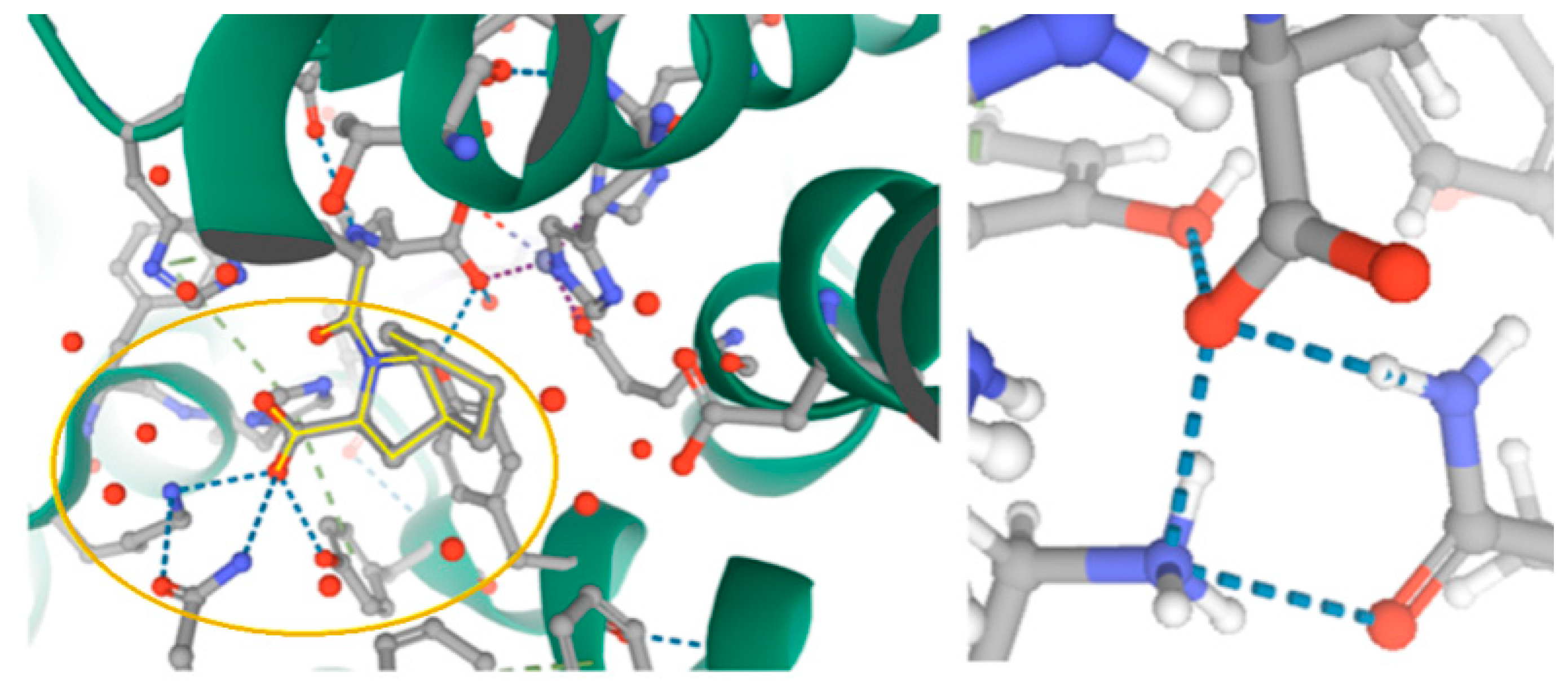
2.2. Further Insight into the ACEI Crystal Structures
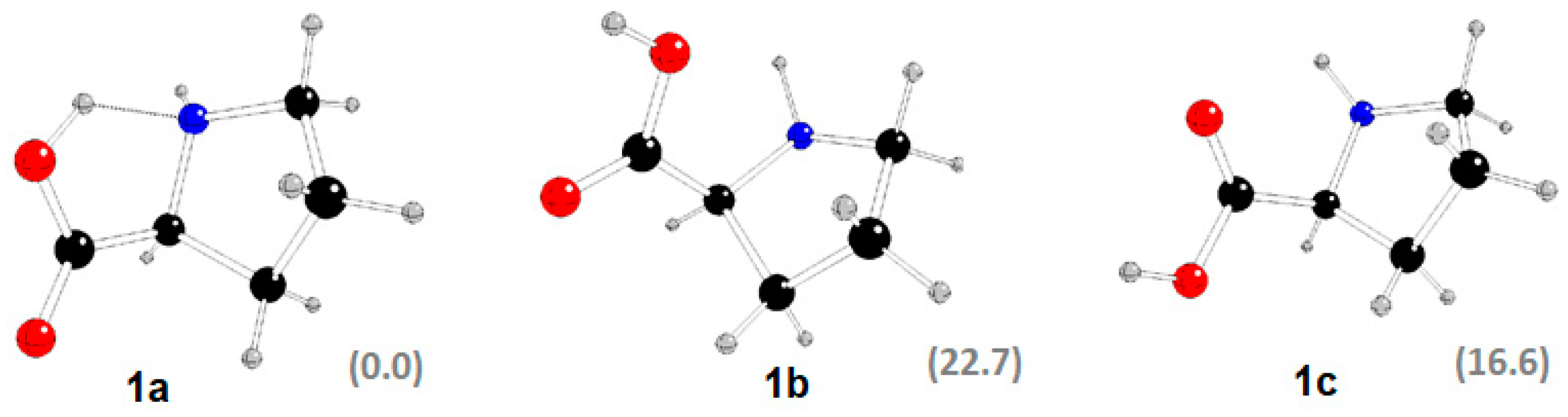
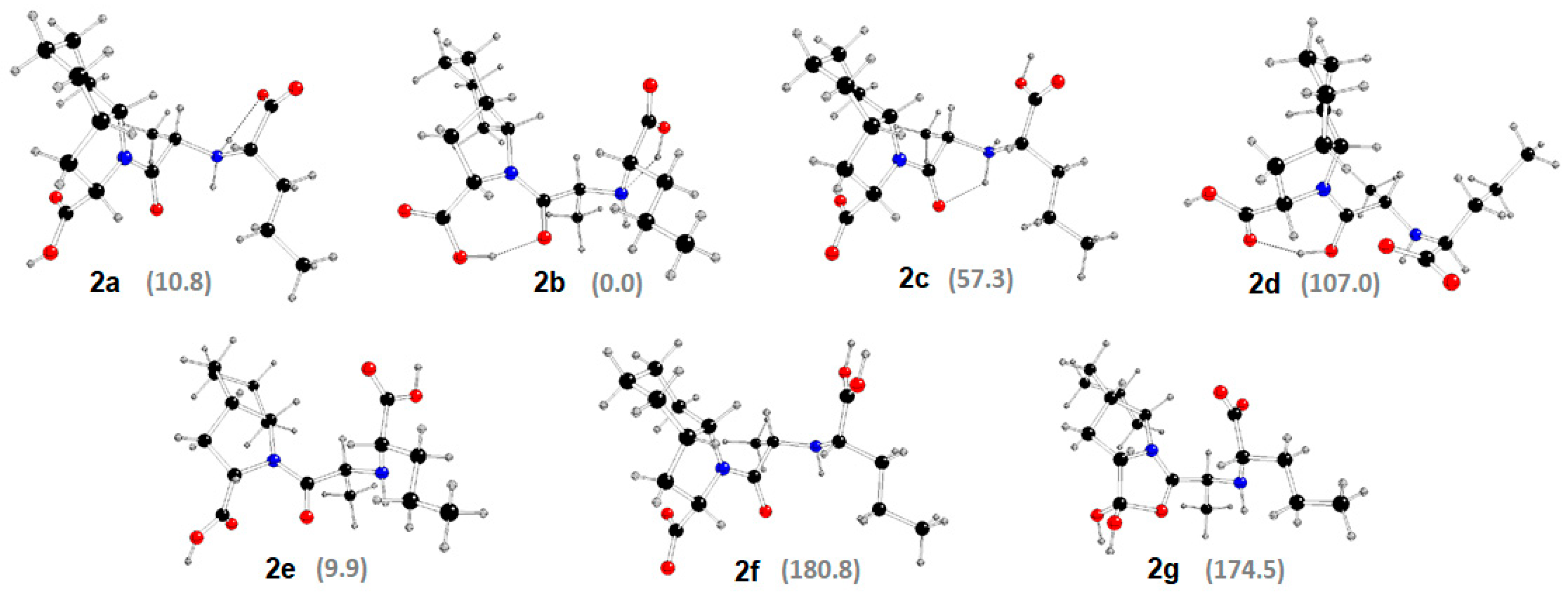
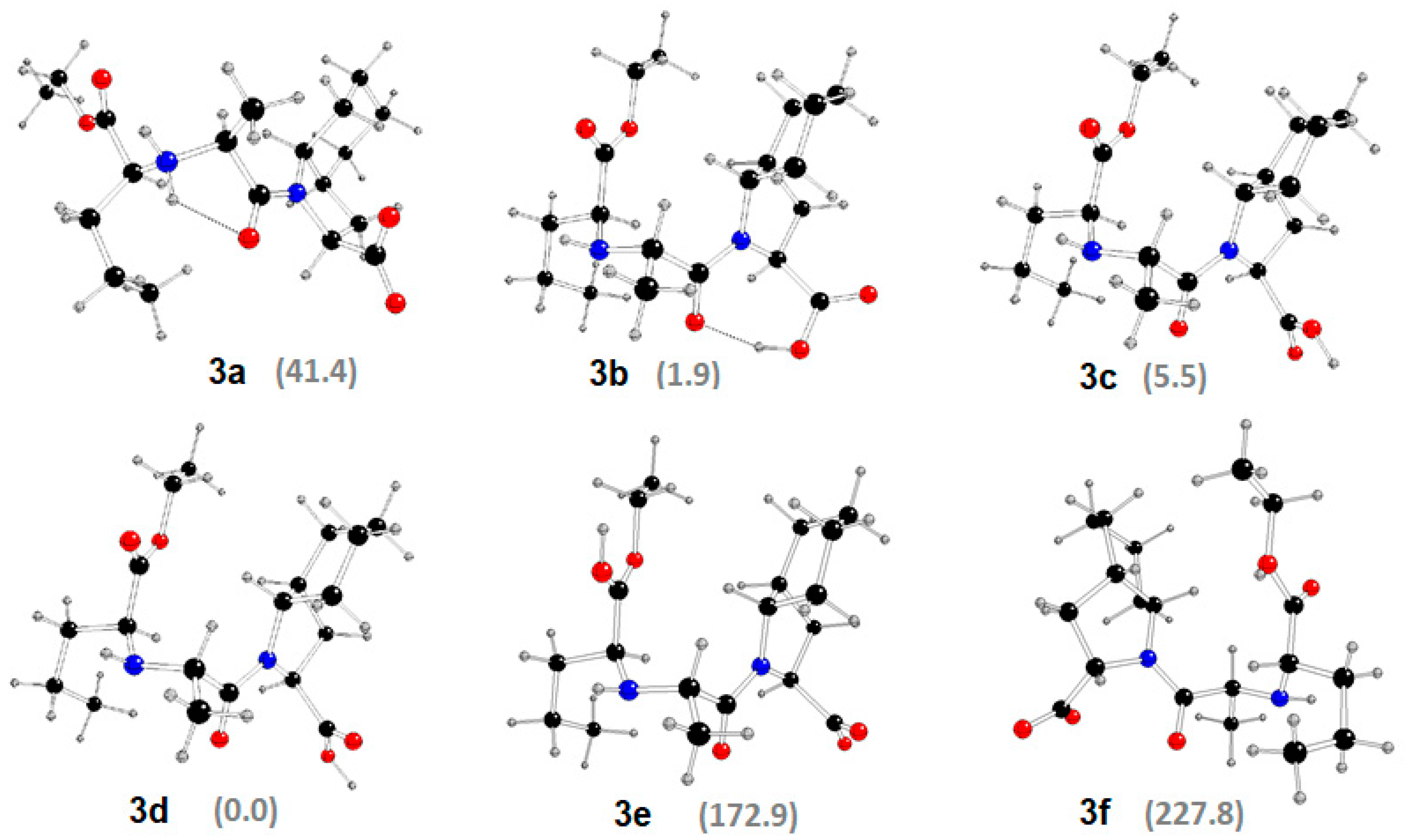
2.3. DFT Study
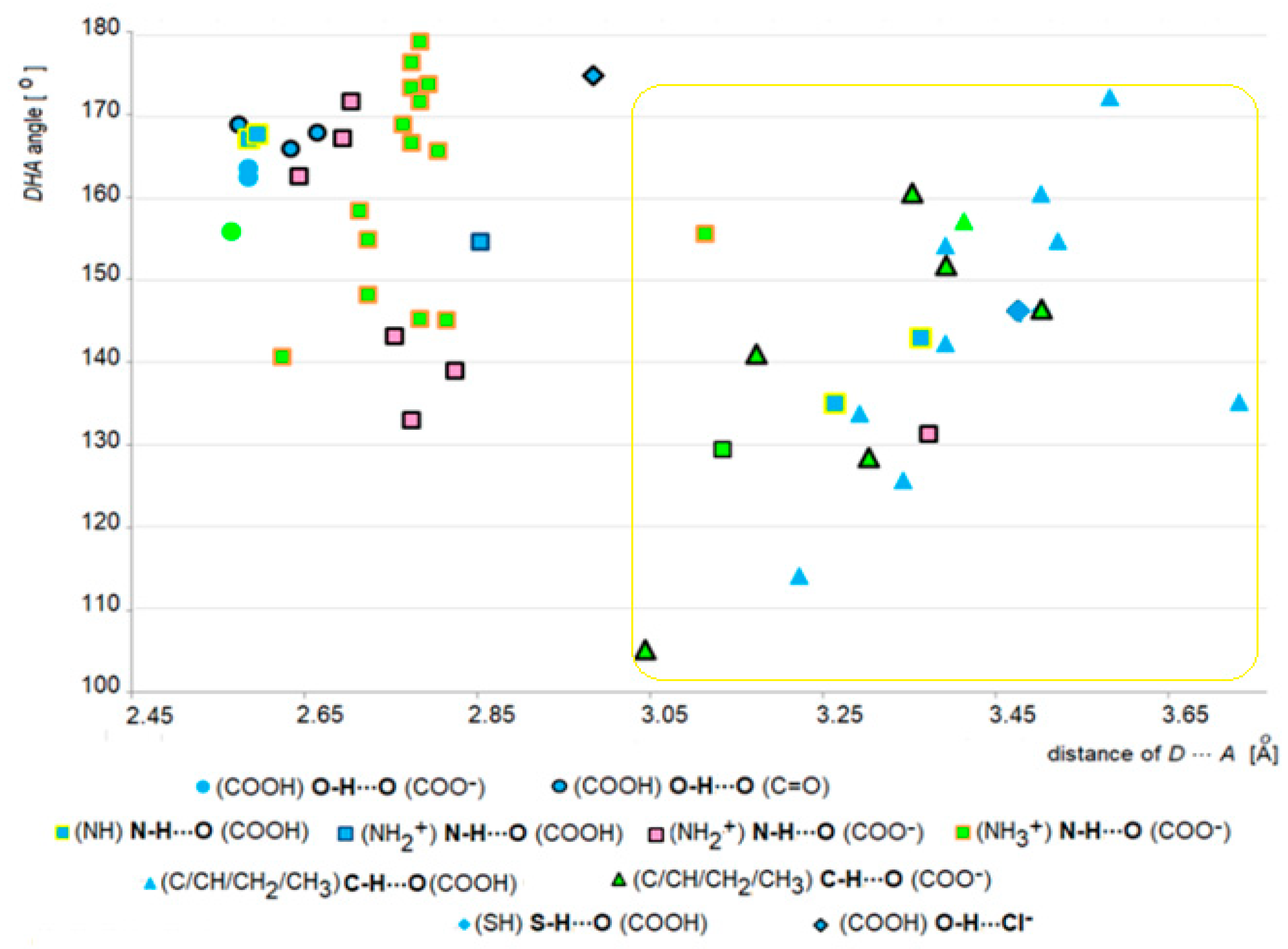
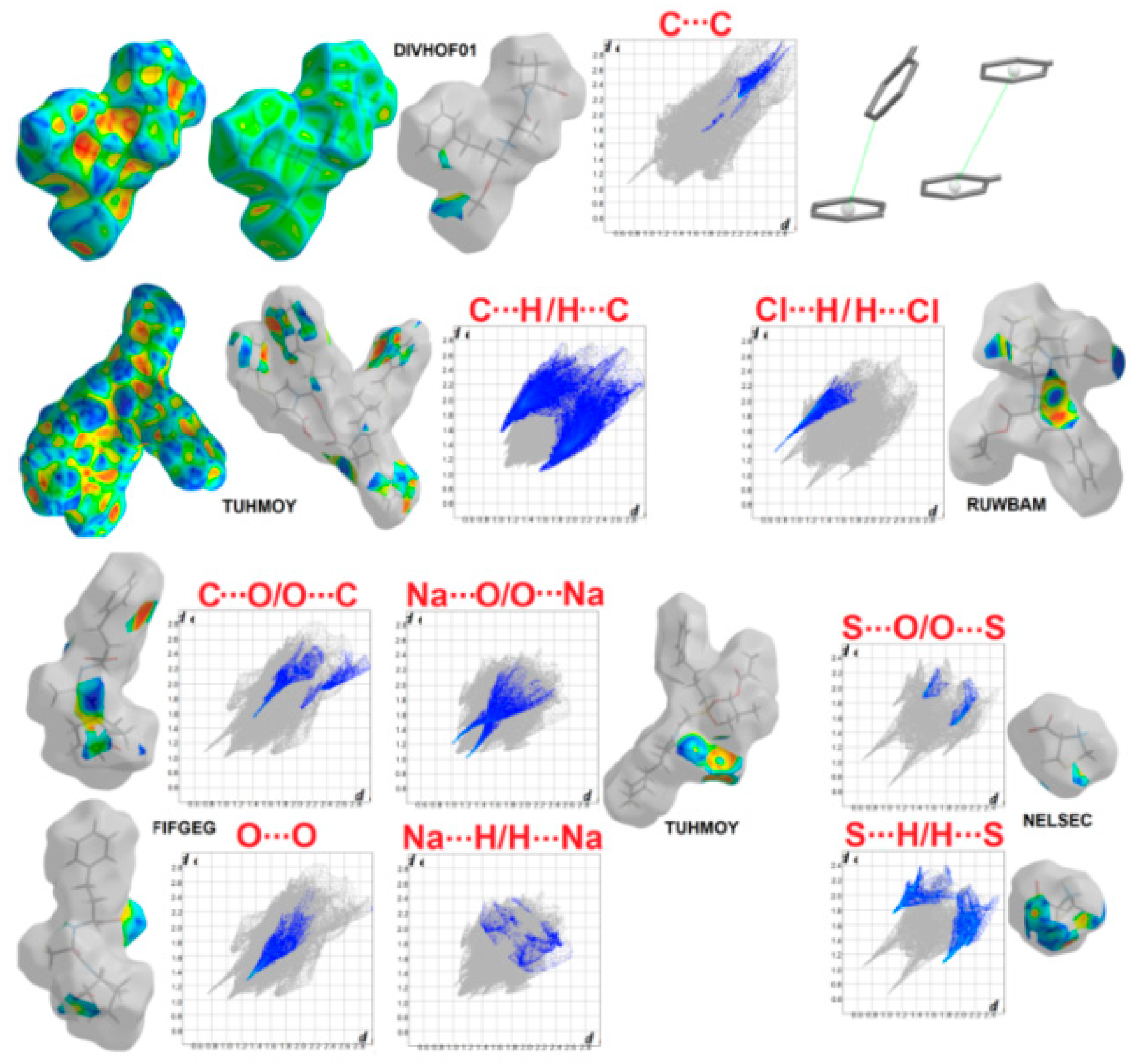
2.4. Survey of Supramolecular Interactions in ACEI Crystal Structures
2.4.1. Hirshfeld Surface Study
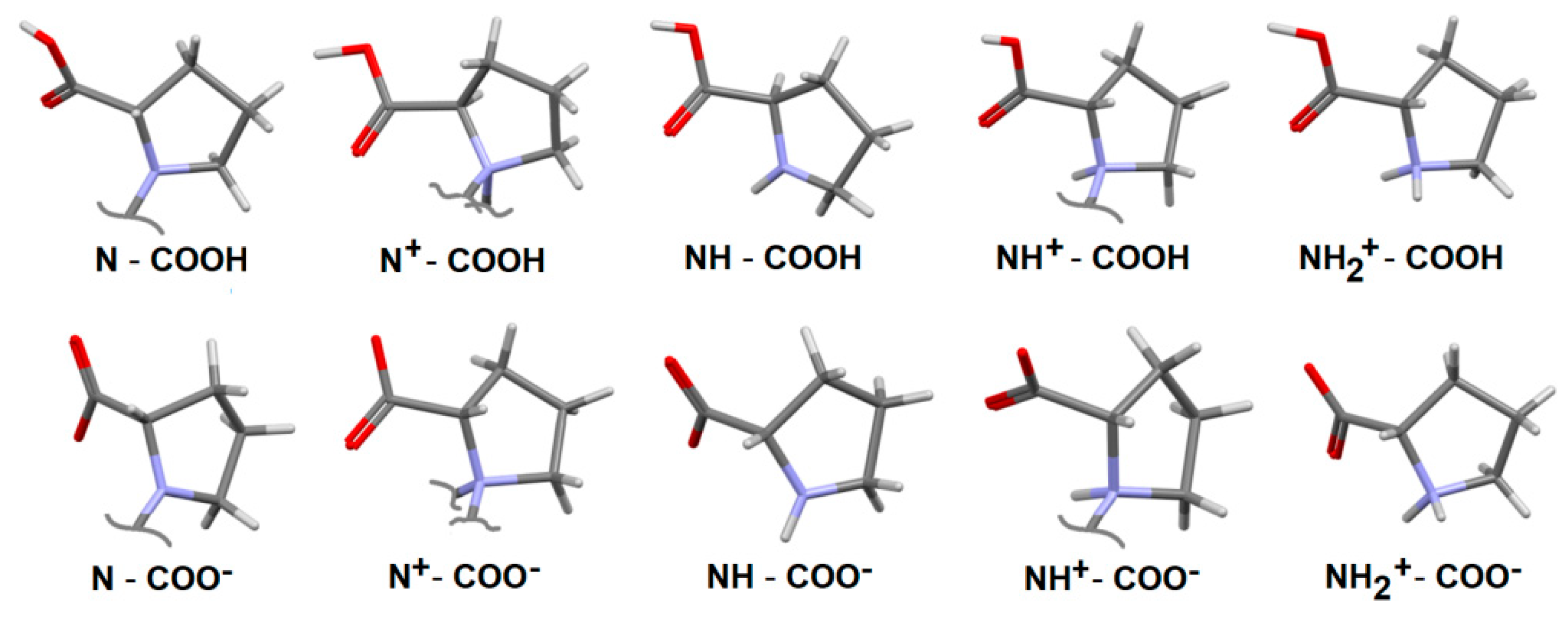
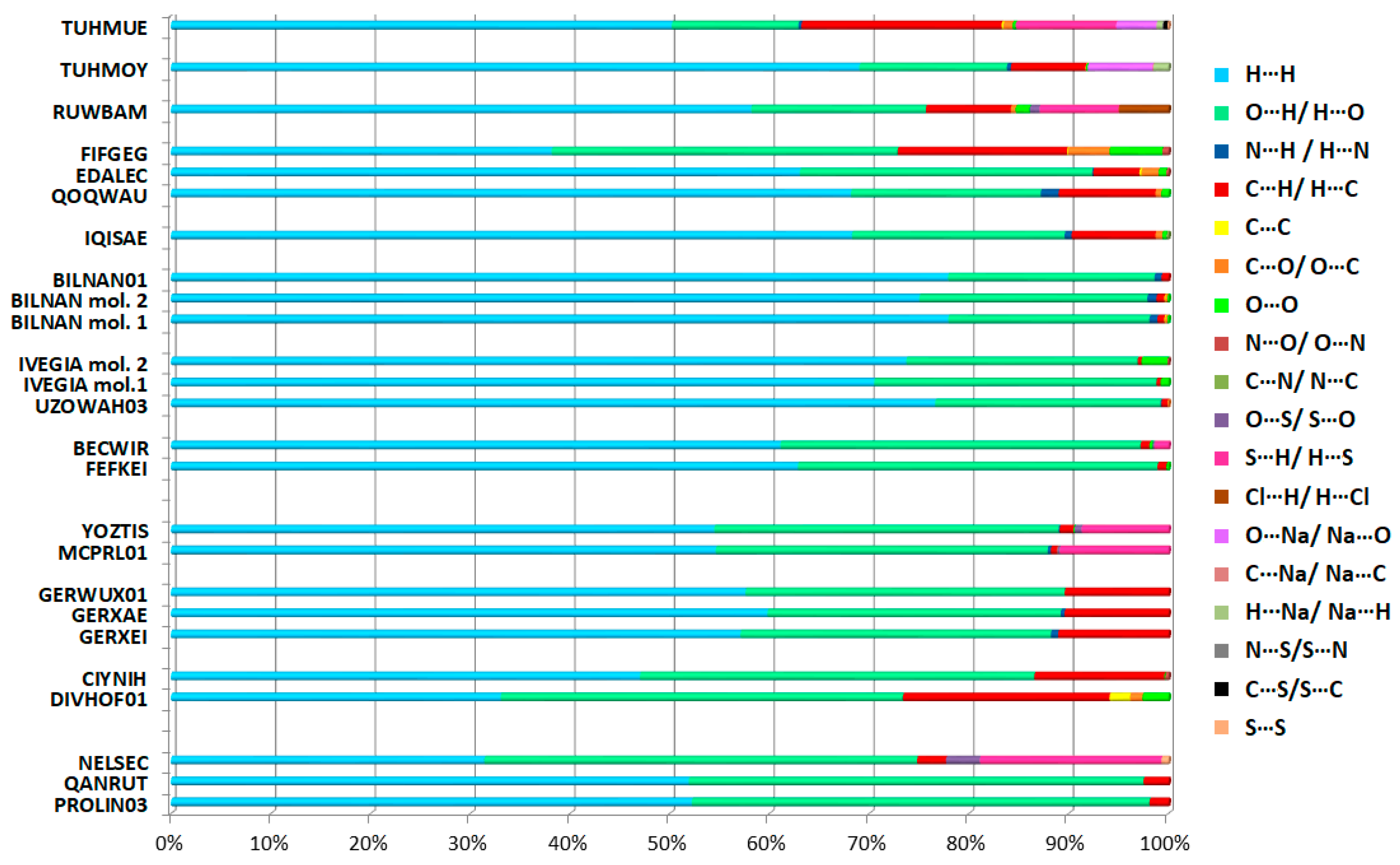
2.4.2. Interaction Enrichment Ratio Analysis
- -
- O…H/H…O in all cases, apart from sodium fosinopril (TUHMOY);
- -
- C…H/H…C: in all proline structures, both perindoprilat forms (BECWIR, FEFKEI) and enalapril maleate (DIVHOF01), enalaprilat (CIYNIH), lisinopril (GERXEI), lisinopril hydrates (GERXAE, GERWUX01), captopril disulfide (YOZTIS), trandolapril (IQISAE), ramipril (QOQWAU), ramiprilat methanol solvate (FIFGEG), spirapril (RUWBAM), sodium fosinopril (TUHMOY), sodium hemi zofenopril (TUHMUE);
- -
- S…H/H…S: in proline analog (NELSEC), perindoprilat DMSO solvate (BECWIR) and YOZTIS, RUWBAM, TUHMUE;
- -
- N…H/H…N: DKP perindopril structure (BILNAN) and QOQWAU;
- -
- C…C: DIVHOF01;
- -
- O…O: perindopril form (IVEGIA);
- -
- C…O/O…C: ramiprilat (EDALEC), RUWBAM;
- -
- Cl…H/H…Cl and S…O/O…S: RUWBAM;
- -
- Na…O/O…Na: TUHMOY, TUHMUE;
- -
- H…H: perindoprilat DMSO solvate (BECWIR), both DKP perindopril forms [BILNAN(01)], perindopril structures and also EDALEC, GERXEI, GERWUX01, TUHMOY, TUHMUE.
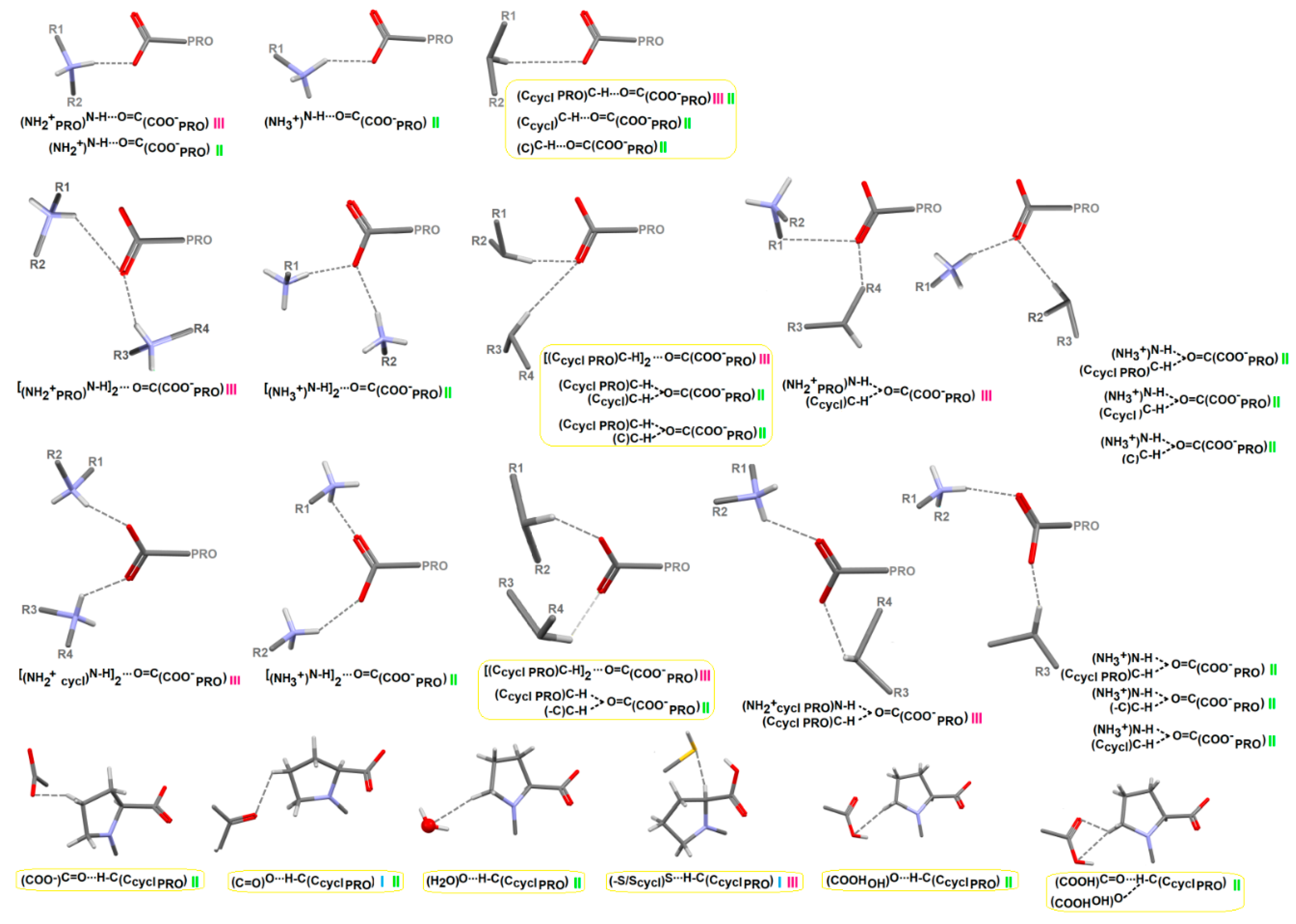
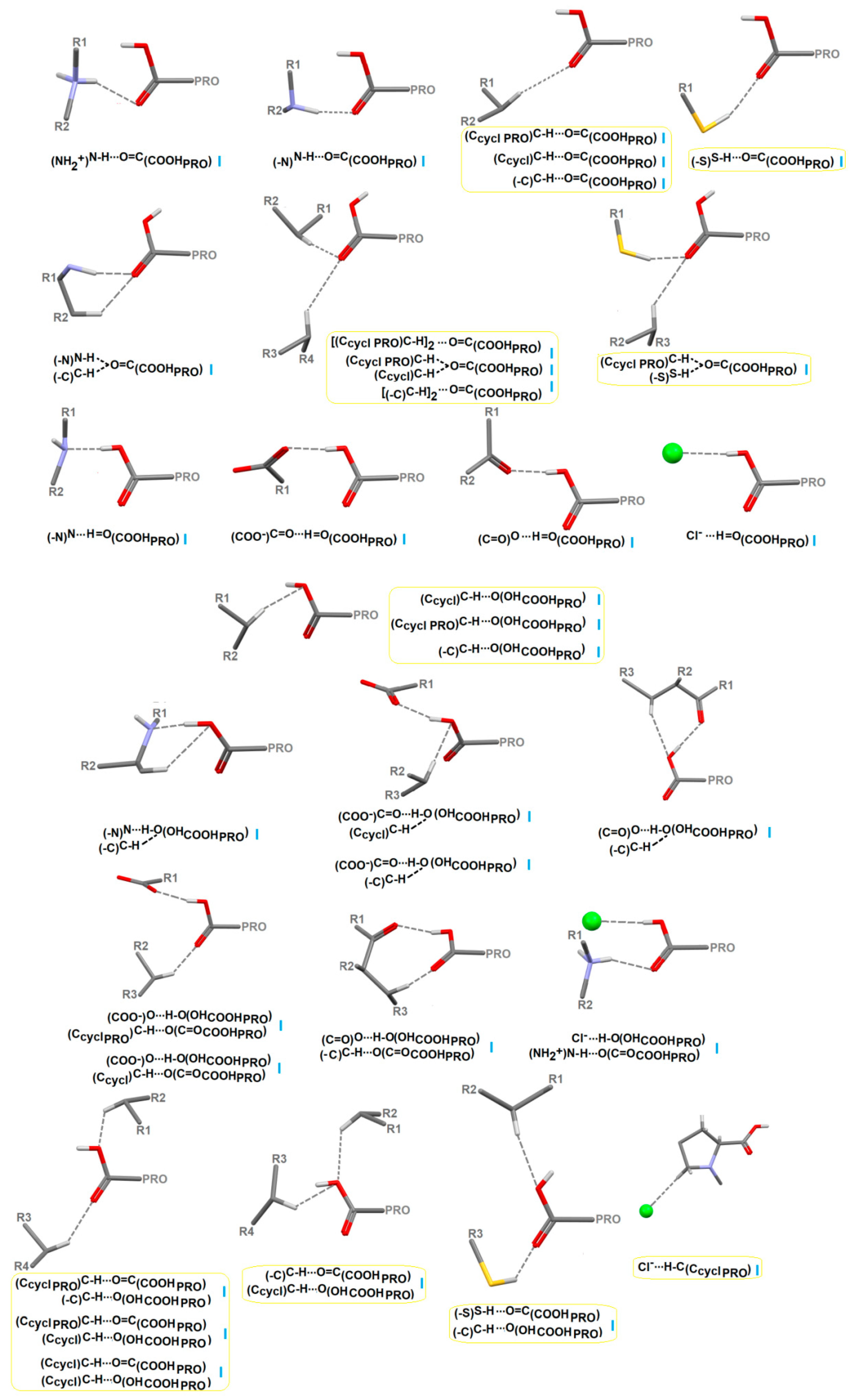
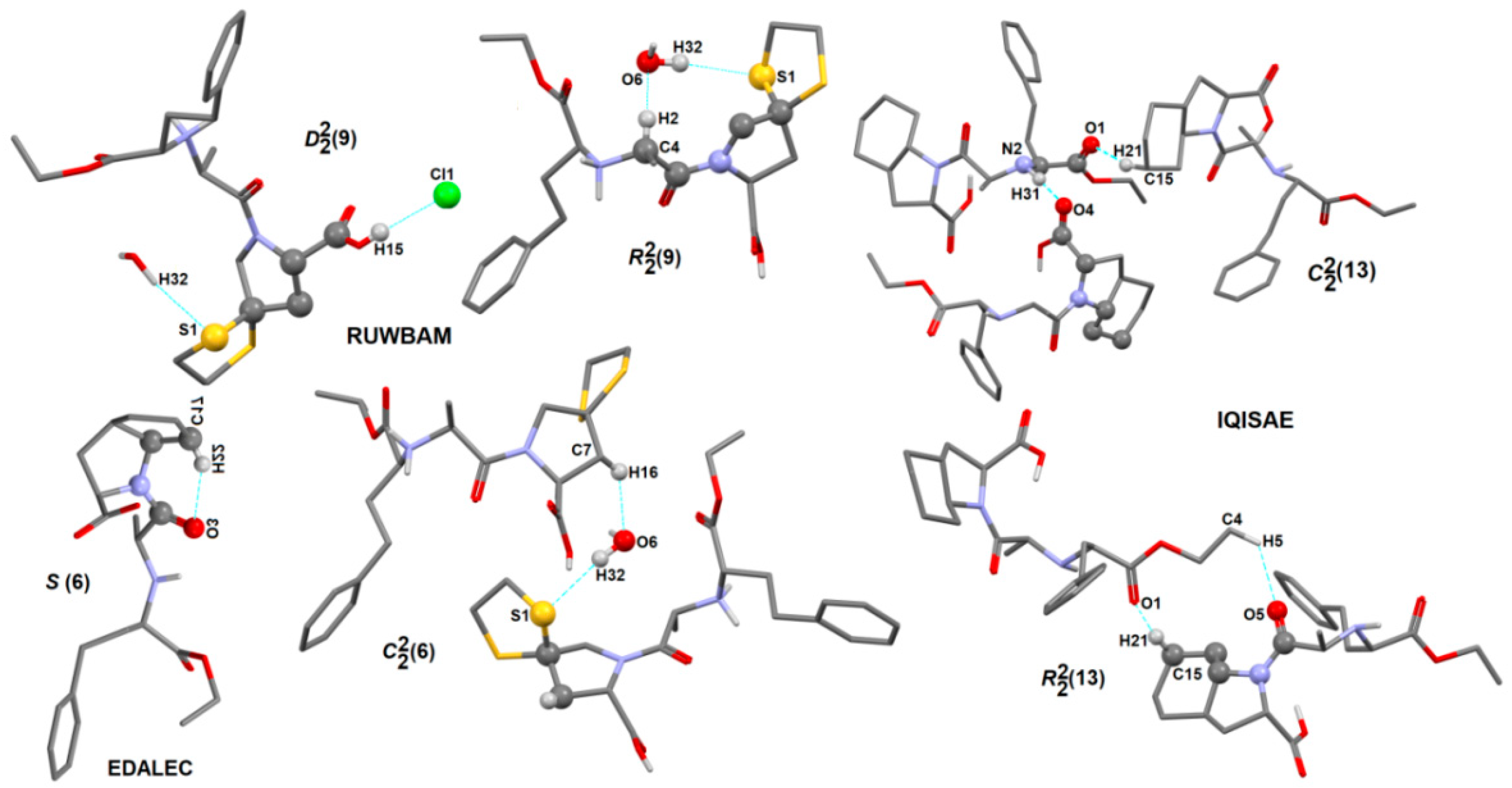
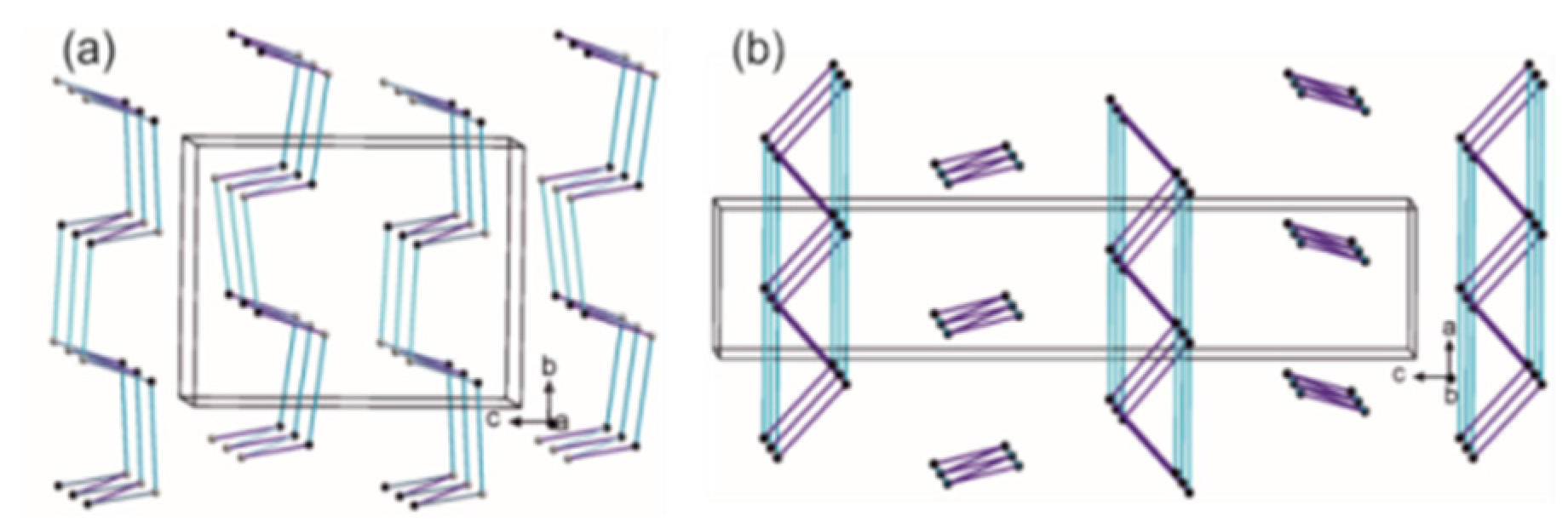
2.4.3. Portfolio of Supramolecular Synthons Resulting from the Proline-Based Tectons
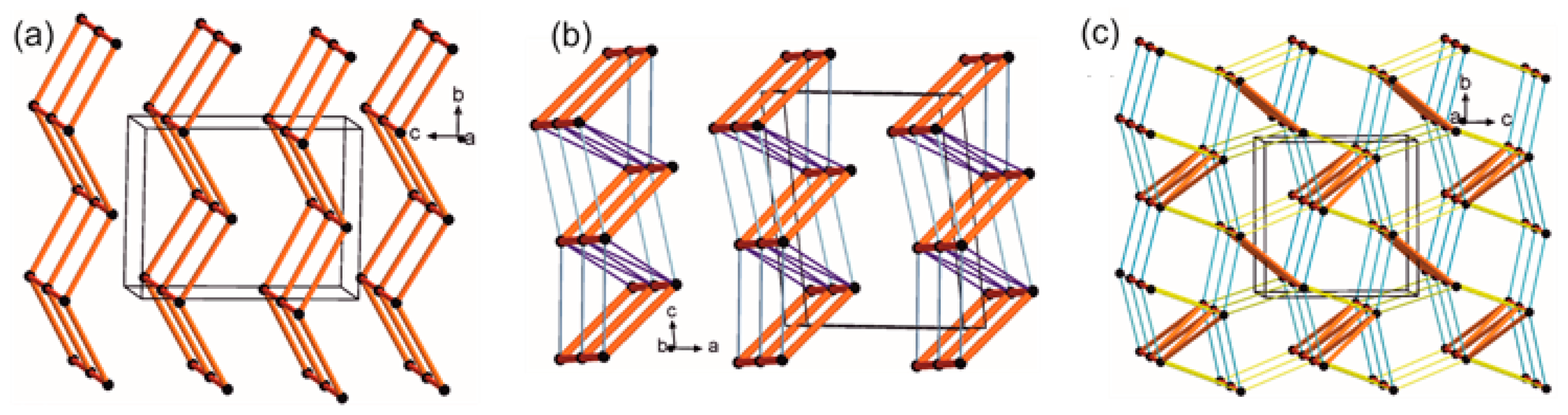
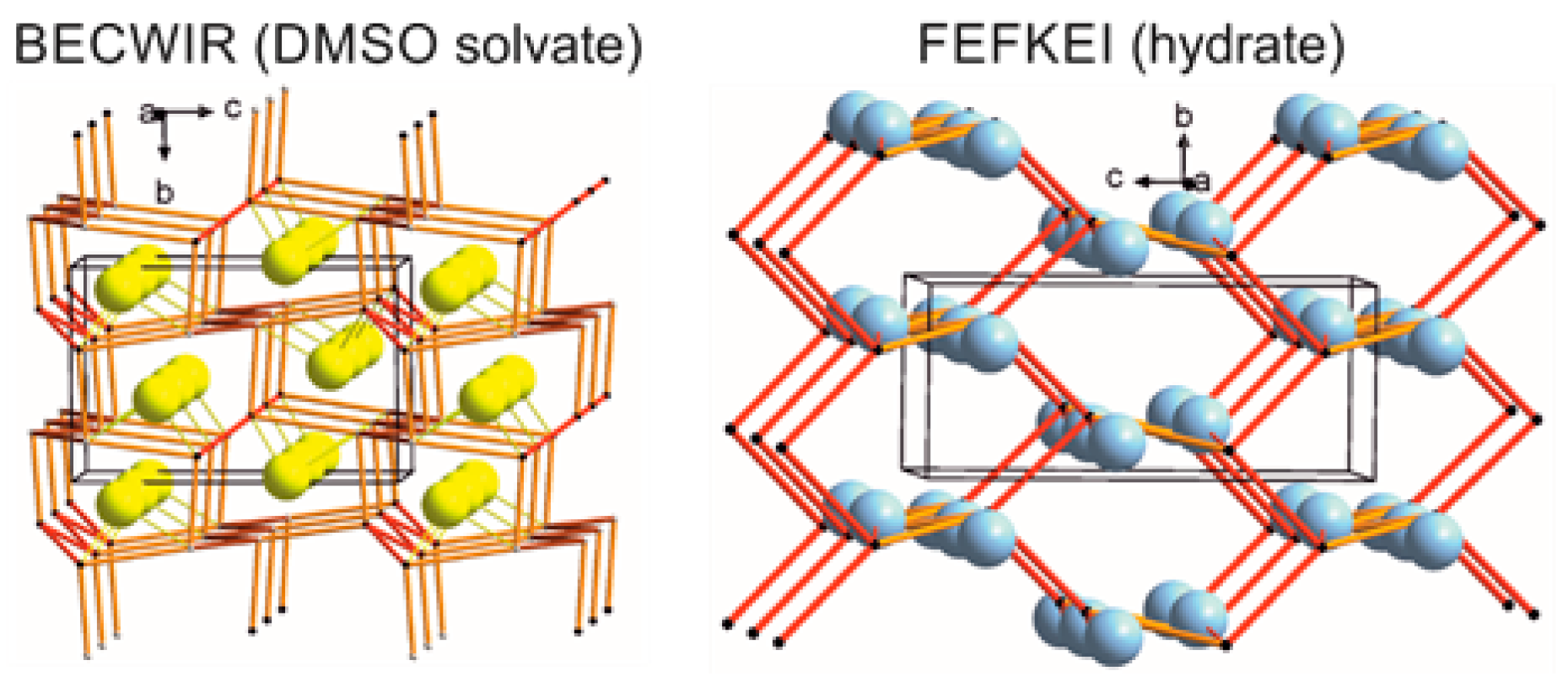
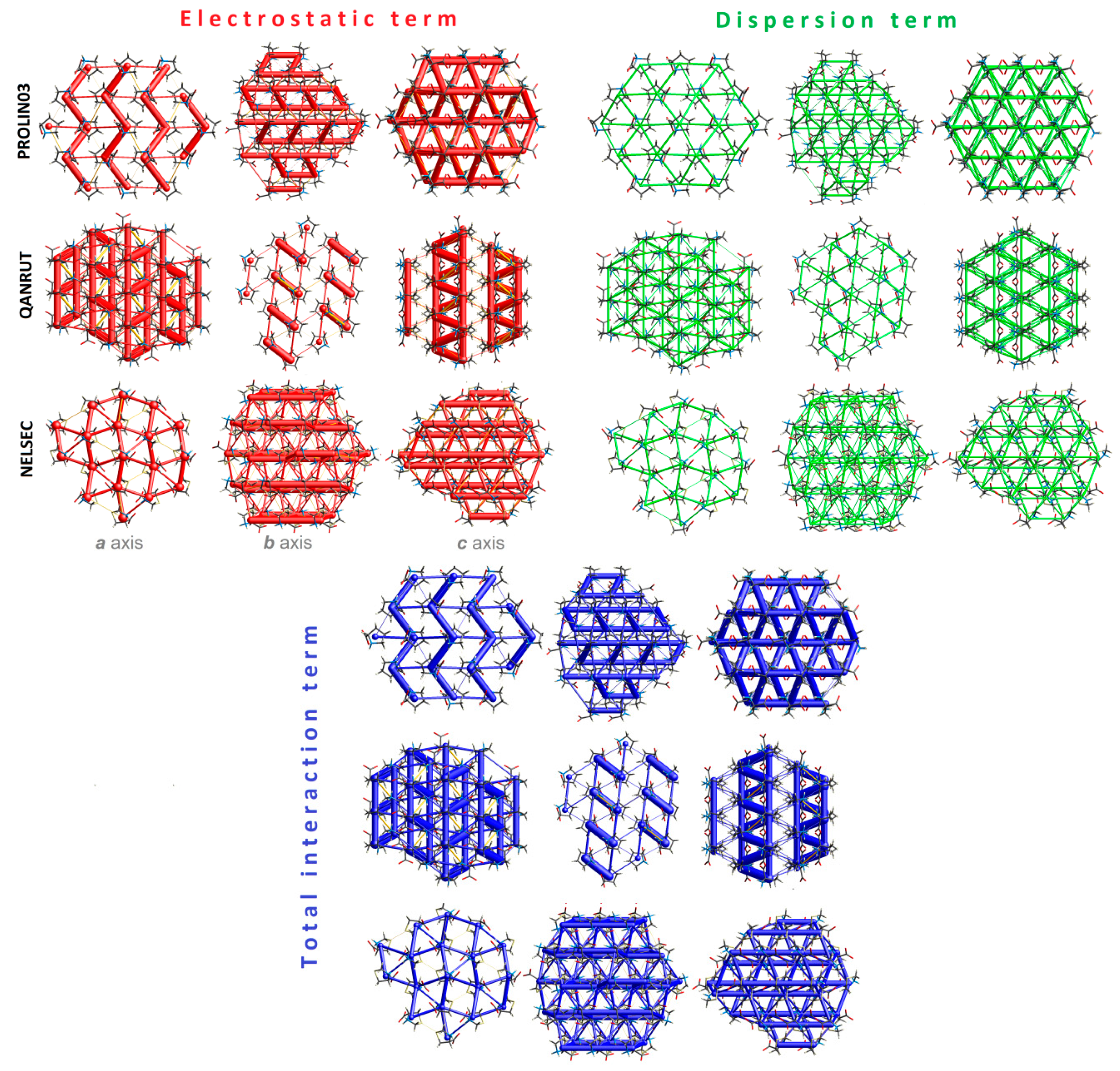
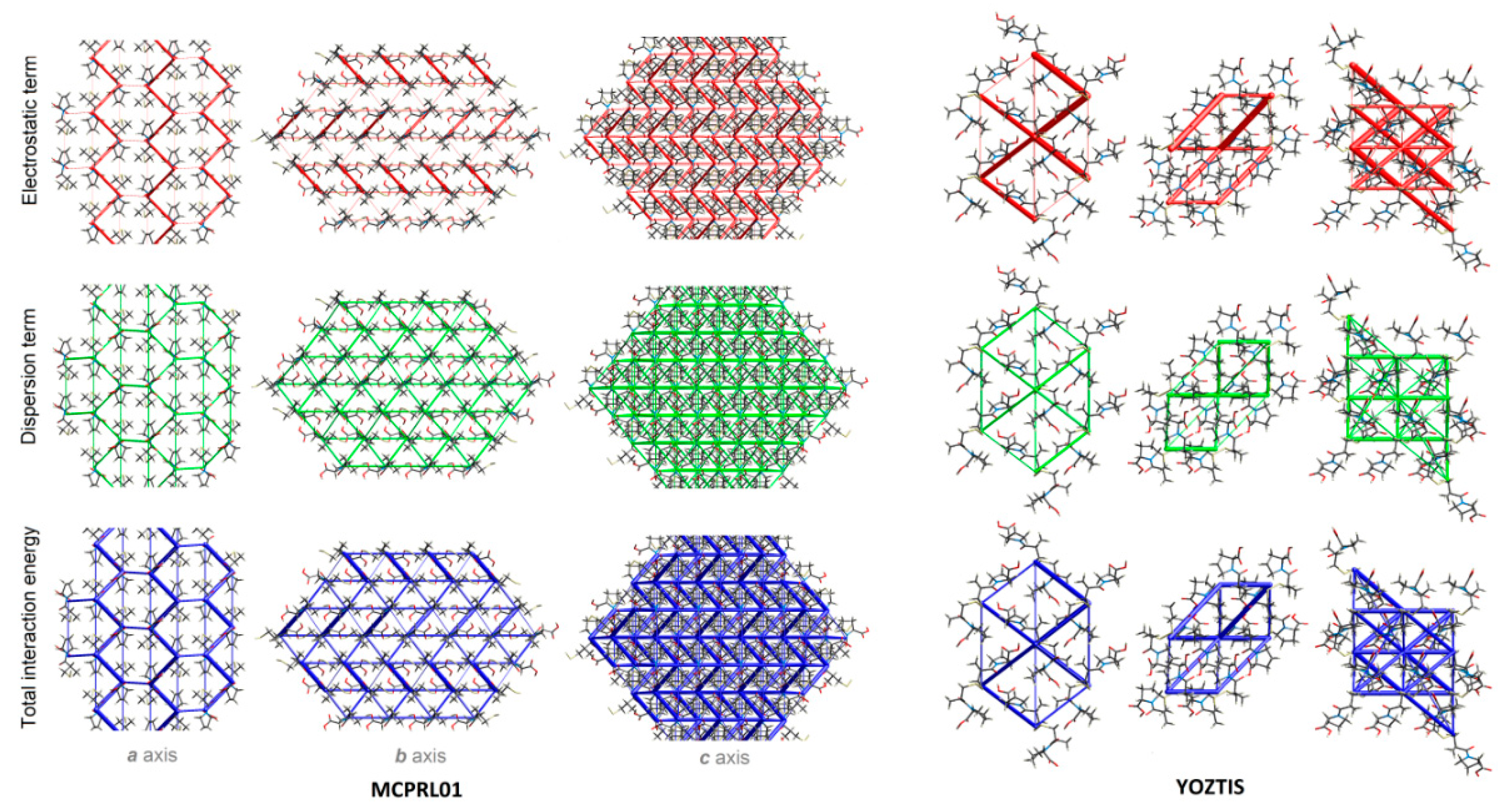
2.4.4. LSAM: Long- Range Synthon Aufbau Modules
Proline Structures
Proline-Based ACEI
Perindoprilat Crystals
Perindopril Erbumine Structures
DKP Perindopril Erbumine Structures
Other Crystal Structures of Other Modified Proline-Based ACEI
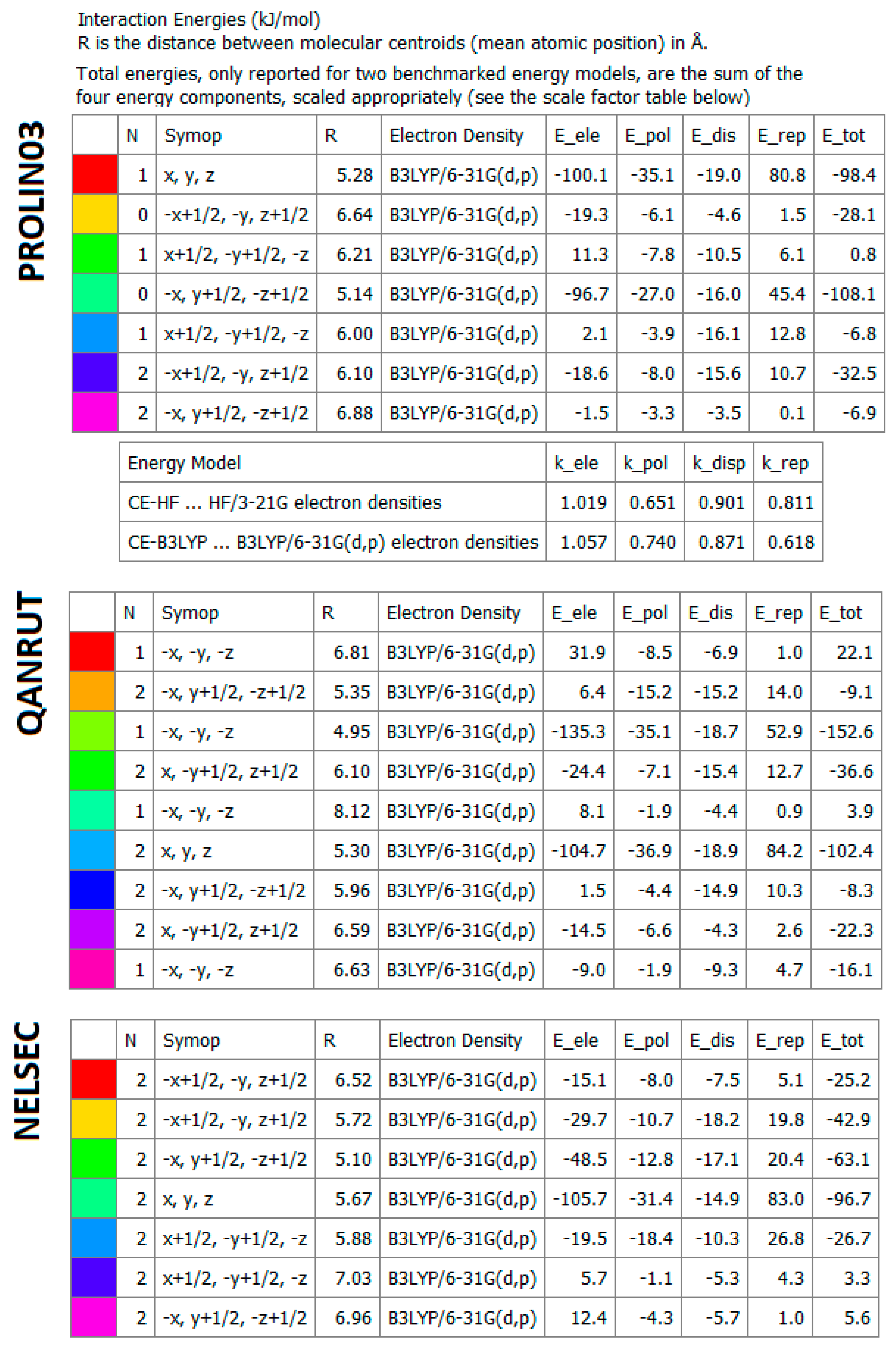
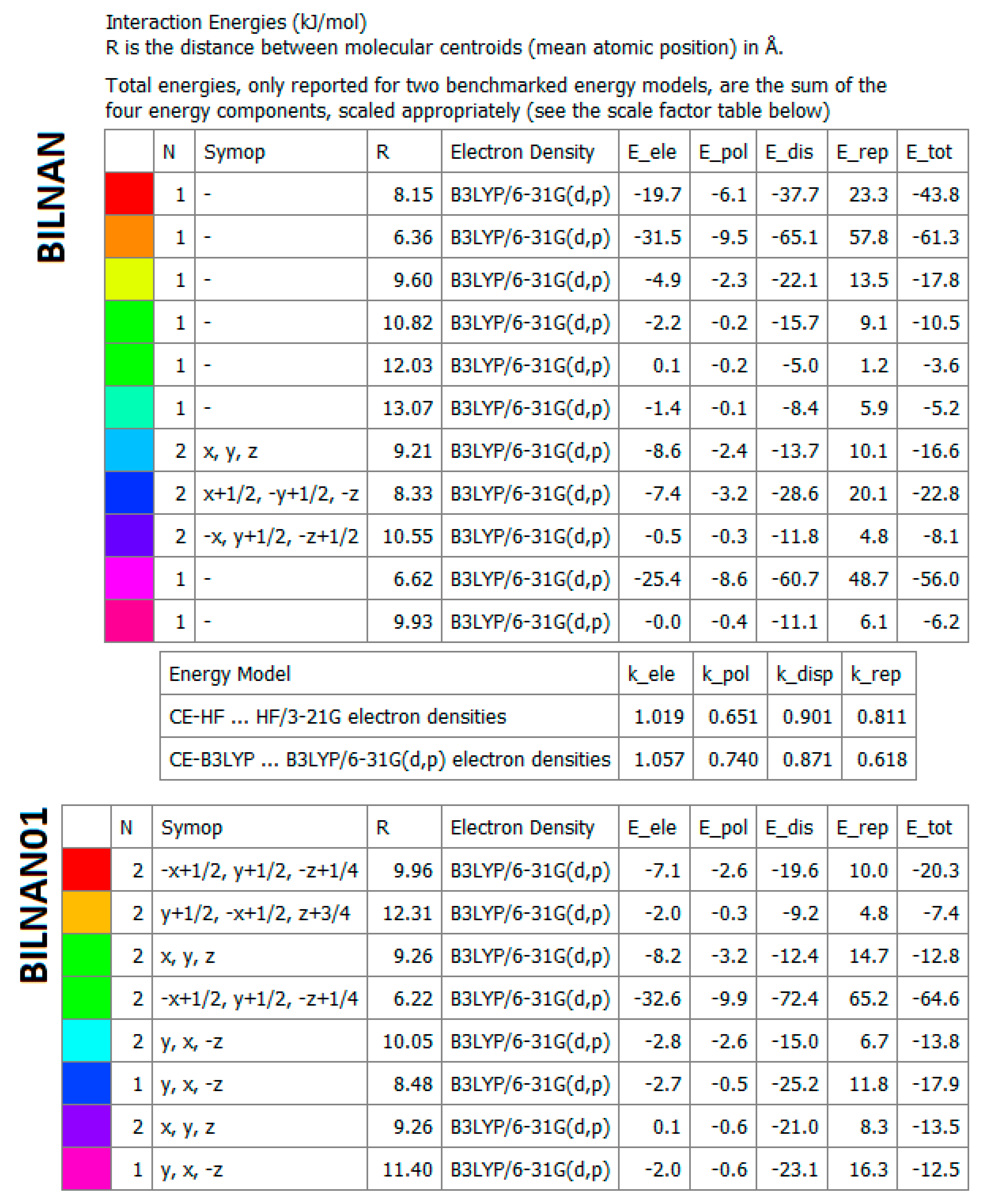
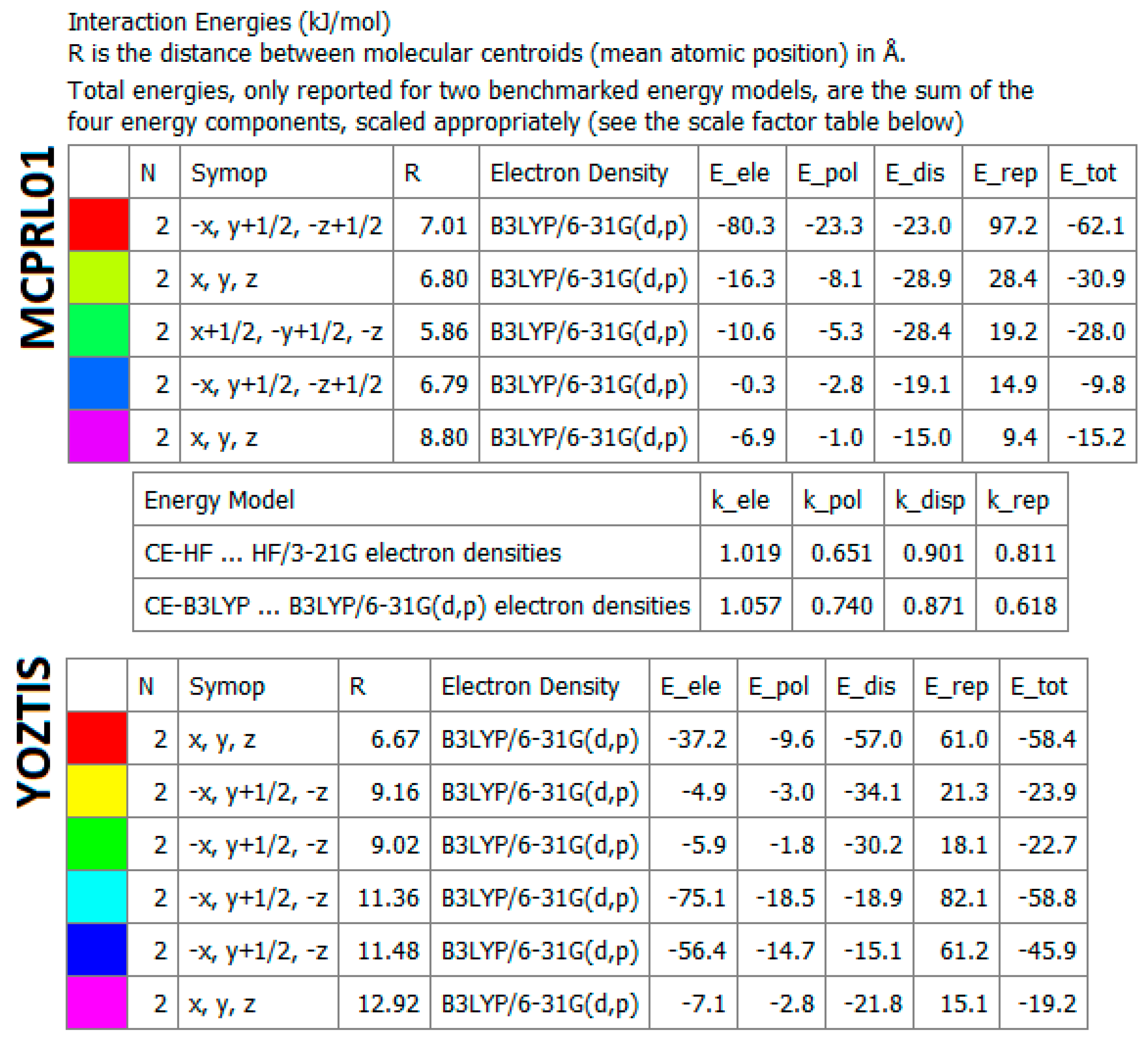
2.4.5. Energy Frameworks on Interaction Energies
2.5. In Silico Control of Pharmacological Profile and Toxicity of ACEI and Their Impurities
3. Materials and Methods
3.1. DFT Studies
3.2. Hirshfeld Surface Analysis and Molecular Electrostatic Potentials
3.3. Enrichment Ratio
3.4. Energy Frameworks
3.5. SwissADMET Profile
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | angiotensin-coverting enzyme |
| ACEI | ACE inhibitors |
| ADMET | absorption, distribution, metabolism, excretion, toxicity |
| BBB | blood-brain barrier |
| CNS | central nervous system |
| CSD | Cambridge Structure Database |
| COX | cycloooxygenase |
| CPCM | conductor like polarizabel continue model |
| 2D | 2-dimensional |
| 3D | 3-dimensional |
| de | distances from the HS to the nearest atom outside the surface |
| DFT | Density Functional Theory |
| di | distances from the HS to the nearest atom inside the surface |
| DKP | diketopiperazine |
| dnorm | normalized contact distance |
| EP | electrostatic potential |
| ER | enrichment ratio |
| FP | fingerprint plot |
| HS | Hirshfeld surface |
| LSAM | Long-range synthon Aufbau modules |
| RCSB PDB | Research Collaboratory for Structural Bioinformatics Protein Data Bank |
| Eele | electrostatic term of energy: |
| Edisp | dispersion term of energy |
| Erep | repulsion term of energy |
| Epol | polarization term of energy |
| Etot | total energy |
| NPA | natural population analysis |
| PSA | polar surface area |
| Vds | |
| vdW | van der Waals radii |
| WHO | World Health Organization |
References
- Stoll, R. From my life. The memoirs of Richard Willstratter. J. Chem. Educ. 1966, 43, A608. [Google Scholar]
- Fischer, G.; Aumuller, T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 2003, 148, 105–150. [Google Scholar] [PubMed]
- Morgan, A.A.; Rubenstein, E. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner-White, J.E.; Bell, L.H.; Maccallum, P.H. Pyrrolidine ring puckering in cis and trans proline residues in proteins and polypeptides.Different puckers are favoured in certain situations. J. Mol. Biol. 1992, 228, 725–734. [Google Scholar] [CrossRef]
- Craveur, P.; Praveen, J.A.; Poulain, P.; Brevern, A.G.; Rebehmed, J. Cis-trans isomerization of omega dihedrals in proteins. Amino Acids 2013, 45, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Mitra, A.K. An explanation for the rare occurrence of cis peptide units in proteins and polypeptides. J. Mol. Biol. 1976, 107, 85–92. [Google Scholar] [CrossRef]
- Berkholz, D.S.; Driggers, C.M.; Shapovalov, M.V.; Dunbrack, R.L.; Karplus, P.A. Nonplanar peptide bonds in proteins are common and conserved but not biased toward active sites. Proc. Natl. Acad. Sci. USA 2012, 109, 449–453. [Google Scholar] [CrossRef] [Green Version]
- De Tar, F.D.; Luthra, N.P. Conformations of proline. J. Am. Chem. Soc. 1977, 99, 1232–1244. [Google Scholar] [CrossRef]
- Altona, C.; Sundaralingman, M. Conformational analysis of the sugar ring in nucleosides and nucleotides. New description using the concept of pseudorotation. J. Am. Chem. Soc. 1972, 94, 8205–8212. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Lakshminarayanan, A.V.; Sabesan, M.N.; Tegoni, G.; Venkatesan, K.; Ramachandran, G.N. Studies on the conformation of amino acids. VI. Conformation of the proline ring as observed in crystal structures of amino acids and peptides. Int. J. Protein Res. 1971, 3, 25–33. [Google Scholar] [CrossRef]
- Némethy, G.; Gibson, K.D.; Palmer, K.A.; Yoon, C.N.; Paterlini, G.; Zagari, A.; Rumsey, S.; Scheraga, H.A. Energy parameters in polypeptides. Improved geometrical parameters and nonbonded interactions for use inECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 1992, 96, 6472–6484. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. Peptidomimetics I; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–25. [Google Scholar]
- Ashida, T.; Kakudo, M. Conformations of prolyl residues in oligopeptides. Bull. Chem. Soc. Jpn. 1974, 47, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, G.N.; Lakshminarayanan, A.V.; Balasubramanium, R.; Tegoni, G. Energy calculations on proline residues. Biochim. Biophys. Acta 1970, 221, 165–181. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Pal, D. Main-chain conformational features at different conformations of sidechains in proteins. Protein Eng. 1998, 11, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, H.K.; Basu, G. Conformational landscape of substituted prolines. Biophys. Rev. 2020, 12, 25–39. [Google Scholar] [CrossRef]
- Pal, D.; Chakrabarti, P. Cis peptide bond in proteins: Residues involved, their confromations, interactions and locations. J. Mol. Biol. 1999, 294, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitagliano, L.; Berisio, R.; Mastrangelo, A.; Mazzarella, L.; Zagari, A. Preferred prolinę puckerings in cis and trans peptide groups: Implications for collagen stability. Protein Sci. 2001, 10, 2627–2632. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef]
- SIB Swiss Institute of Bioinformatics Members The SIB Swiss Institute of Bioinformatics‘ resources: Focus on curated databases. Nucleic Acids Res. 2016, 44, D27–D37. [CrossRef]
- Pandey, A.K.; Naduthambi, D.; Thomas, K.M.; Zondlo, N.J. Proline editing: A general and practical approach to the synthesis of functionally and structurally diverse peptides. Analysis of steric versus stereoelectronic effects of 4-substituted prolines on conformation within peptides. J. Am. Chem. Soc. 2013, 135, 4333–4363. [Google Scholar] [CrossRef] [Green Version]
- Lenci, E.; Trabocchi, A. Occurence of the D-proline chemotype in enzyme inhibitors. Symmetry 2019, 11, 558. [Google Scholar] [CrossRef] [Green Version]
- Christgen, S.L.; Becker, D.F. Role of proline in pathogen and host interactions. Antioxid. Redox Signal 2019, 30, 683–709. [Google Scholar] [CrossRef]
- De Luca, L.; Chiminazzo, A.; Sperni, L.; Strukul, G.; Scarso, A. Pyrrolidine-Containing Bisphosphonates as Potential Anti-Resorption Bone. Drugs. Chem. A Eur. J. 2017, 23, 3474–3478. [Google Scholar] [CrossRef] [Green Version]
- Trapero, A.; Llebaria, A. A prospect for pyrrolidine iminosugars as antidiabetic glucosidase inhibitors. J. Med. Chem. 2012, 55, 10345–10346. [Google Scholar] [CrossRef]
- Vitali, A. Proline-rich peptides: Multifunctional bioactive molecules as new potential therapeutic drugs. Curr. Protein Pept. Sci. 2015, 16, 147–162. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Hu, C. Chiral Drugs: An Overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Singh, K.; Shakya, P.; Kumar, A.; Alok, S.; Kamal, M.; Prakash, S. Stereochemistry and its role in drug design. IJPSR 2014, 5, 4644–4659. [Google Scholar]
- Olczak, A.; Główka, M.; Szczesio, M.; Bojarska, J.; Duax, W.; Burkhart, B.; Wawrzak, Z. Nonstoichiometric complex of gramicidin D with KI at 0.8 A loisresolution. Acta Cryst. D 2007, 63, 319–327. [Google Scholar]
- Olczak, A.; Główka, M.; Bojarska, J.; Szczesio, M.; Wawrzak, Z.; Duax, W.L. The first crystal structure of a gramicidin complex with sodium: High-resolution study of a nonstoichiometric gramicidin D-NaI complex. Acta Cryst. D 2010, 66, 874–880. [Google Scholar]
- Główka, M.; Olczak, A.; Bojarska, J.; Szczesio, M. Structural puzzles of complexed gramicidins in their crystals. Wiadomości Chem. 2007, 61, 161–187. [Google Scholar]
- Główka, M.; Olczak, A.; Bojarska, J.; Szczesio, M.; Duax, W.; Burhart, B.; Pangborn, W.; Langs, D.; Wawrzak, Z. Structure ofgramicidin D-RbCl complex at atomic resolution from low-temperature synchrotron data: Interactions of double-strandedgramicidin channel contents and cations with channel wall. Acta Cryst. D 2005, 61, 433–441. [Google Scholar]
- Główka, M.; Olczak, A.; Bojarska, J.; Szczesio, M.; Duax, W.; Burkhart, B.; Pangborn, W.; Langs, D.; Li, N.; Wawrzak, Z. Ion channels in crystals of gramicidin D complex with RbCl. Atomic resolution low-temperature synchrotron X-ray data. Acta Crystallogr. Sect. A Found. Crystallogr. 2004, 60, 165. [Google Scholar]
- Görbitz, C.H. Crystal structures of amino acids: From bond lenghts in glycine to metal complexes and high-prrssure polymorphs. Crystallogr. Rev. 2015, 21, 160–212. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Bahra, G.; Brown, C.R.; Hitchcock, P.B.; Patell, Y.; Seddon, K. L-Proline 2, 5-dihydroxybenzoic acid (1/1): A zwitterion co-crystal. Acta Chem. Scand. 1995, 49, 762–767. [Google Scholar] [CrossRef]
- Tilborg, A.; Norberg, B.; Wouters, J. Pharmaceutical salts and cocrystals involving amino acids: A brief structural overview of the state-of-art. Eur. J. Med. Chem. 2014, 74, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.S.; Seddigi, Z.S.; Bajee, S.; Azeeza, S.; Riyaz, S.; Ahmed, S.A.; Althagafi, I.I.; Jamal, Q.M.S.; Kamal, A. Multicomponent access to novel proline/cyclized cysteine tethered monastrol conjugates as potential anticancer agents. J. Saudi Chem. Soc. 2019, 23, 503–513. [Google Scholar] [CrossRef]
- Tumanova, N.; Tumanov, N.; Robeyns, K.; Fischer, F.; Fusaro, L.; Morelle, F.; Ban, V.; Hautier, G.; Filinchuk, Y.; Wouters, J.; et al. Opening Pandora’s Box: Chirality, Polymorphism, and Stoichiometric Diversity in Flurbiprofen/Proline Cocrystals. Cryst. Growth Des. 2018, 18, 954–961. [Google Scholar] [CrossRef]
- Salaman, P.; Ramon, C. Co-Crystals of Celecoxib and L-Proline. Patent EP2325172A1, 25 May 2011. [Google Scholar]
- Pandey, J.; Prajapati, M.R.; Shimpi, P.; Tandon, S.P.; Velaga, A.; Srivastava, A.; Sinha, K. Studies of molecular structure, hydrogen bonding and chemical activity of a nitrofurantoin-L-proline cocrystal: A combined spectroscopic and quantum chemical approach. RSC Adv. 2016, 6, 74135–74154. [Google Scholar] [CrossRef]
- Yadav, B.; Balasubramanian, S.; Chavan, R.B.; Thipparaboina, R.; Naidu, V.G.M.; Shastri, N.R. Hepatoprotective cocrystals and salts of riluzole: Synthesis, solid-staet characterization, and evaluation. Cryst. Gworth Des. 2018, 18, 1047–1061. [Google Scholar] [CrossRef]
- He, H.; Huang, Y.; Zhang, Q.; Wang, J.R.; Mei, X. Zwitterionic cocrystals of flavonoids and proline: Solid-state characterization, pharmaceutical properties, and pharmacokinetic performance. Cryst. Growth Des. 2016, 16, 2348–2356. [Google Scholar] [CrossRef]
- Liu, M.; Hong, C.; Yao, Y.; Shen, H.; Ji, G.; Li, G.; Xie, Y. Development of a pharmaceutical cocrystal with solution crystallization technology: Preparation, characterization, and evaluation of myricetin-proline cocrystals. Eur. J. Pharm. Biopharm. 2016, 107, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. Cryst. Eng. Commun. 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Srinivasan, V.; Balasubramanian, D. Proline is a Protein-Compatible Hydrotrope. Langmuir 1995, 11, 2830–2833. [Google Scholar] [CrossRef]
- Wang, L.Y.; Yu, Y.M.; Jiang, F.B.; Li, Y.T.; Wu, Z.Y.; Yan, C.W. The first zwiteterionic cocrystal of indomethacin with amino acid showing optimized physicochemical properties as well as accelerated absorption and slowed elimination in vivo. New J. Chem. 2020, 44, 3930–3939. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Sasisekharan, V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968, 23, 283–437. [Google Scholar]
- Brandts, J.F.; Halvorson, H.R.; Brennan, M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 1975, 14, 4953–4963. [Google Scholar] [CrossRef]
- Babul, J.; Nakagawa, A.; Stellwagen, E. An examination of the involvement of proline peptide isomerization in protein folding. J. Mol. Biol. 1978, 126, 117–121. [Google Scholar] [CrossRef]
- Schmid, F.X.; Grafl, R.; Wrba, A.; Beintema, J.J. Role of proline peptide bond isomerisation in unfolding and refolding of ribonuclease. Proc. Natl. Acad. Sci. USA 1986, 83, 872–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.E.; Sarkar, A.; Wampler, J.E. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 1990, 214, 253–260. [Google Scholar] [CrossRef]
- MacArthur, M.W.; Thornton, J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991, 218, 397–412. [Google Scholar] [CrossRef]
- Frommel, C.; Preissner, R. Prediction of prolyl residues in cis-conformation in protein structures on the basis of the amino acid sequence. FEBS 1990, 277, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Pahlke, D.; Leitner, D.; Wiedemann, U.; Labudde, D. COPS-cis/trans peptide bond confromation prediction of amino acids on the basis of secondary structure information. Struct. Bioinform. 2005, 21, 685–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exarchos, K.P.; Exarchos, T.P.; Papaloukas, C.; Troganis, A.N.; Fotiadis, D.I. PBOND: Web server for the prediction of proline and non-proline cis/trans isomerization. Genom. Proteom. Bioinform. 2009, 7, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Joosten, R.P.; Salzemann, J.; Bloch, V.; Stockinger, H.; Berglund, A.C.; Blanchet, C.; Bongcam-Rudloff, E.; Combet, C.; Da Costa, A.L.; Deleage, G.; et al. PDB_REDO: Automated re-refinement of X-ray structure models in the PDB. J. Appl. Cryst. 2009, 42, 376–384. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Jossten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Bartlett, G.J.; Woolfson, D.N. On the satisfaction of bckbone-carbonyl lone pairs of electrons in protein structures. Protein Sci. 2016, 25, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Pan, X.M. The contribution of prolinę residues to protein stability is associated with isomerization equilibrium in both unfolded and folded states. Extremophiles 2009, 13, 481–489. [Google Scholar] [CrossRef]
- Weiss, M.S.; Jabs, A.; Hilgenfeld, R. Peptide bonds revisited. Nat. Struct. Biol. 1998, 5, 676. [Google Scholar] [CrossRef]
- Williams, C.J.; Videau, L.L.; Hintze, B.J.; Richardson, D.C.; Richardson, J.S. Cis-nonPro peptides: Genuine occurences and their functional roles. bioRxiv 2018, 324517. [Google Scholar]
- Fischer, S.; Dunbrack, R.L., Jr.; Karplus, M. Cis-Trans Imide Isomerization of the Proline Dipeptide. J. Am. Chem. Soc. 1994, 116, 11931–11937. [Google Scholar] [CrossRef]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, H. Molecular Dynamics Simulations Towards the Understanding of the Cis-Trans Isomerization of Proline as a Conformational Switch for the Regulation of Biological Processes. Dissert Thesis, Georgia Univeristy, Athens, GA, USA, 2014. [Google Scholar]
- Shinoda, K.; Fujitani, H. Initiation of prolyl cis-trans isomerisationin the CDR-H3 loop of an antibody in response to antigenbinding. Sci. Rep. 2017, 7, 16964–16976. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, W.J.; Welekr, E.; Scheraga, H.A. Proline cis-trans isomerization and protein folding. Biochemistry 2002, 41, 14637–14644. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef] [Green Version]
- Borgia, A.; Kemplen, K.R.; Borgia, M.B.; Soranno, A.; Shammas, S.; Wunderlich, B.; Nettels, D.; Best, R.B.; Clarke, J.; Schuler, B. Transient misfolding dominates multidomain protein folding. Nat. Commun. 2015, 6, 8861–8871. [Google Scholar] [CrossRef] [Green Version]
- Comes, S.; Gagliardi, M.; Laprano, N.; Fico, A.; Cimmino, A.; Palamidessi, A.; De Cesare, D.; De Falco, S.; Angelini, C.; Scita, G.; et al. L-proline induces a mesenchymal-like invasive program in embroynic stem cells by remodelling H3K9 and H3K36 methylation. Stem. Cell Rep. 2013, 1, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.A.; Ytreberg, F.M. The cis conformation of prolinę leads to weaker binding of a p53 peptide to MDM2 compared to trans. Arch. Biochem. Biophys. 2015, 575, 22–29. [Google Scholar] [CrossRef]
- Schulz, G.E.; Schirmer, R.H. Principles of Protein Structure; Springer: New York, NY, USA, 1979; pp. 17–26. [Google Scholar]
- Baldwin, R.L. The search for folding intermediates and the mechanism of protein folding. Ann. Rev. Biophys. 2008, 37, 1–21. [Google Scholar] [CrossRef]
- Levitt, M. Effect of proline residues on protein folding. J. Mol. Biol. 1981, 145, 251–263. [Google Scholar] [CrossRef]
- Brazin, K.N.; Mallis, R.J.; Fulton, D.B.; Andreotti, A.H. Reg-ulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclo-philin A. Proc. Natl. Acad. Sci. USA 2002, 99, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.; Reichman, C.; Saleh, T.; Birge, R.; Kalodimos, C. Proline cis-trans isomerization controls autoinhibition of a signalling protein. Mol. Cell 2007, 25, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Wulf, G.; Finn, G.; Suizu, F.; Lu, K. Phosphorylation-specific prolyl isomerization: Is there an underlying theme? Nat. Cell Biol. 2005, 7, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Lummis, S.; Beebe, D.; Lee, L.; Lester, H.; Broadhurst, R.; Dougherty, D. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.; Santos-Rosa, H.; Kouzarides, T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 2006, 126, 905–909. [Google Scholar] [CrossRef] [Green Version]
- Santiveri, C.M.; Perez-Canadillas, J.M.; Vadivelu, M.K.; Allen, M.D.; Rutherford, T.J.; Watkins, N.A.; Bycroft, M. NMR structureof the alpha-hemoglobin stabilizing protein: Insights into conforma-tional heterogeneity and binding. J. Biol. Chem. 2004, 279, 34963–34970. [Google Scholar] [CrossRef] [Green Version]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two confor-mational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Yao, J.F.; Feng, S.Z.; He, Y.; Jiang, E.L.; Zhang, R.L.; Yang, D.L.; Gong, M.; Zheng, X.H.; Chen, S.L. BCR-ABL enhances the prolylisomerase activity of Pin 1 by interacting with DAPK1 in ph+ ALL. Cancer Med. 2018, 7, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancerand a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef]
- Schmid, X.; Mayr, L.M.; Mucke, M.; Schonbrunner, E.R. Prolyl iso-merases: Role in protein folding. Adv. Protein Chem. 1993, 44, 25–66. [Google Scholar]
- Fischer, G. Chemical aspects of peptide bond isomerization. Chem. Soc. Rev. 2000, 29, 119–127. [Google Scholar] [CrossRef]
- Dugave, C.; Demange, L. Cis–trans isomerization of organic mole-cules and biomolecules: Implications and applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Sun, A.; Lu, P.J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.H.; Li, X. The prolyl isomerase Pin1 reg-ulates amyloid precursor protein processing and amyloid-beta pro-duction. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Torbeev, V.Y.; Hilvert, D. Both the cis-trans equilibrium and isomerization dynamics of a single proline amide modulate β2-microglobulin amyloid assembly. Proc. Natl. Acad. Sci. USA 2013, 110, 20051–20200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siltari, A.; Viitanen, R.; Kukkurainen, S.; Vapaatalo, H.; Valjakka, J. Does the cis/trans configuration of peptide bonds in bioactive tripeptides play a role in ACE-I enzyme inhibition? Biolocs Targets Ther. 2014, 8, 59–65. [Google Scholar]
- Nollet, L.M.; Toldra, F. Handbook of Analysis of Active Compounds in Functional Foods; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Gomez-Ruiz, J.A.; Recio, I.; Belloque, J. ACE-inhibitory activity and structural properties of peptide Asp-Lys-Ile-Pro-[beta-CN f(47–51)]. Study of the peptide forms synthesized by different methods. J. Agric. Food Chem. 2004, 52, 6315–6319. [Google Scholar] [CrossRef]
- Bouabdallah, S.; Trabelsi, H.; Dhia, M.T.B.; Hamida, N.B. Kinetic study on the isomerization of perindopril by HPLC. Chromatographia 2012, 75, 1247–1255. [Google Scholar] [CrossRef]
- Bouabdallah, S.; Dhia, M.T.B.; Driss, M.R. Study of a conformational equilibrium of lisinoperil by HPLC, NMR, DFT. Int. J. Anal. Chem. 2014, 494719. [Google Scholar]
- Bursell, E. The role of proline in energy metabolism. In Energy Metabolism in Insects; Downer, R., Ed.; Springer: Boston, MA, USA, 1981; pp. 135–154. [Google Scholar]
- Zhang, L.; Alfano, J.R.; Becker, D.F. Proline metabolism increases katG expression and oxidative stress resistance in Escherichia coli. J. Bacteriol. 2015, 197, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Moses, S.; Sinner, T.; Zaprasis, A.; Stoveken, N.; Hoffmann, T.; Belitsky, B.R.; Sonenshein, A.L.; Bremer, E. Proline utilization by Bacillus subtilis: Uptake and catabolism. J. Bacteriol. 2012, 194, 745–758. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M. Proline metabolism in cel regulation and cancer biology: Recent advances and hypotheses. Antioxid. Redox Signal 2019, 30, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.Y.L.; Zareba, I.; Baszanowska, W.; Lewoniewska, S.; Palka, J. Understanding the role of key amino acids in regulation of prolinę dehydrogenase/prolinę oxidase (prodh/pox)-dependendent apoptosis/autophagy as an approach to targeted cancer therapy. Mol. Cell. Biochem. 2020, 466, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Navarro, M.; Teixido, M.; Giralt, E. Jumping hurdles: Peptides able to overcome biological barriers. Acc. Chem. Res. 2017, 50, 1847–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Nuchprapha, A.; Paisansak, S.; Sangtanoo, P.; Srimongkol, P.; Saisavoey, T.; Reamtong, O.; Choowongkomon, K.; Karnchanatat, A. Two novel ACE inhibitory peptides isolated from longan seeds: Purification, inhibitory kinetics and mechanisms. RSC Adv. 2020, 10, 12711–12720. [Google Scholar] [CrossRef] [Green Version]
- Chavan, K.; Kedar, N.A. Synthesis, Molecular Modelling and Biological Evaluations of Novel Trans-4-Amino-Proline Derivatives As Potential Cyclooxygenase-2 (Cox-2) Inhibitors. In Proceedings of the International Conference on Drug Discovery (ICDD), Hyderabad, India, 29 February–2 March 2020. [Google Scholar]
- Hussain, Z.; Cooke, A.J.; Neelamkavil, S.; Brown, L.; Carswell, E.; Geissler, W.M.; Guo, Z.; Hawes, B.; Kelly, T.M.; Kiyoi, Y.; et al. Design and synthesis of novel proline based factor XIa selective inhibitors as leads for potential new anticoagulants. Bioorg. Med. Chem. Lett. 2020, 30, 127072. [Google Scholar] [CrossRef] [PubMed]
- Millies, B.; von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Goppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-based allosteric inhibitors of Zika and Dengue virus NS2B/NS3 proteases. J. Med. Chem. 2019, 62, 24. [Google Scholar] [CrossRef]
- Awatef, O.; Neifar, M.; Ouertani, R.; Saleheddine, A.; Mosbah, A.; Cherif, A. Effectiveness of enzyme inhibitors in biomedicine and pharmacotherapy. Adv. Tissue Eng. Regen. Med. 2019, 5, 85–90. [Google Scholar]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bąk, A.; Kaureova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef] [Green Version]
- Fani, N.; Bordbar, A.K.; Ghayeb, Y.; Sepehri, S. Integrating docking and molecular dynamics approaches for a series of proline-based 2,5-diketopiperazines as novel alpha,beta-tubulin inhibitors. J. Biomol. Struct. Dyn. 2015, 33, 2285–2295. [Google Scholar] [CrossRef]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Huang, R.M.; Yi, X.X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.H. An update on 2,5-Diketopiperazines from marine organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Cacciatore, I.; Arslan, M.E.; Fornasari, E.; Marinelli, L.; Di Stefano, A.; Mardinoglu, A. Histydyl-proline diketopierazine isomeres as multipotent anti-Alzheimer drug candidates. Biomolecules 2020, 10, 737. [Google Scholar] [CrossRef]
- Gonzalez, B.D.; Jacobsen, P.B. Depression in lung cancer patients: The role of perceived stigma. Psycho-Oncology 2012, 21, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Cornacchia, C.; Cacciatore, I.; Baldassarre, L.; Mollica, A.; Feliciani, F.; Pinnen, F. 2,5-diketopiperazines as neuroprotective agents. Mini-Rev. Med. Chem. 2012, 12, 2–12. [Google Scholar] [CrossRef]
- Ressurreição, A.S.M.; Delatouche, R.; Gennari, C.; Piarulli, U. Bifunctional 2,5-Diketopiperazines as Rigid Three-Dimensional Scaffolds in Receptors and Peptidomimetics. Eur. J. Org. Chem. 2011, 2011, 217–228. [Google Scholar]
- Ianzer, D. Do the cardiovascular effects of angiotensin-converting enzyme (ACE) I involve ACE-independent mechanisms? New insights from proline-rich peptides of Bothrops jararaca. J. Pharmacol. Exp. Ther. 2007, 322, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Bhuyan, B.J.; Mugesh, G. Effect of peptide-based captopril analogues on angiotensyn converting enzyme activity and peroxynitrite-mediated tyrosine nitration. Org. Biomol. Chem. 2011, 9, 5185–5192. [Google Scholar] [CrossRef]
- Sun, H.; Li, T.J.; Zhao, X.H. ACE inhibition and enzymatic resistance in vivo of a casein hydrolysate subjected to plastein reaction in the presence of extrinsic proline and ethanol or methanol water fractionation. Int. J. Food Prop. 2014, 17, 386–398. [Google Scholar] [CrossRef]
- Manoharan, S.; Shuib, A.S.; Abdullah, N. Structural characteristics and antihypertensive effects of angiotensin-I-converting enzyme inhibitory peptides in the renin-angiotensin and kallikreinginin systems. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 383–406. [Google Scholar] [CrossRef]
- Fan, H.; Liao, W.; Wu, J. Molecular interactions, bioavailability, and cellular mechanisms of ACE inhibitory peptides. J. Food Biochem. 2019, 43, e12572–e12580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuyer, G.; Schwager, S.; Sturrock, E.; Isaac, R.; Acharya, K. Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci. Rep. 2012, 2, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Bentham, J.; Di Cesare, M.; Bixby, H.; Danaei, G.; Cowan, M.J. Worldwide trends in blood pressure from 1975 to 2015: A pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet 2017, 389, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M. Food-Originating ACE inhibitors, including antihypertensive peptides, as preventive food components in blood pressure reduction. Compr. Rev. Food Sci. Food Saf. 2014, 13, 114–134. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E.; Collins, K.J.; Himmejfarb, C.D.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Hypertension 2018, 71, 1269–1324. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abaiobir, A. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Galardy, R.E. Inhibition of angiotensin converting enzymeby phosphoramidates and polyphosphates. Biochemistry 1982, 21, 5777–5781. [Google Scholar] [CrossRef]
- Cushman, D.W.; Pluscec, J.; Williams, N.J.; Weaver, E.R.; Sabo, E.F.; Kocy, O.; Cheung, H.S.; Ondetti, M.A. Inhibition of angiotensin-coverting enzyme by analogs of peptides from Bothrops jararaca venom. Experientia 1973, 29, 1032–1035. [Google Scholar] [CrossRef]
- Ferreira, S.H. Angiotensin converting enzyme: History and relevance. Semin. Perinatol. 2000, 24, 7–10. [Google Scholar] [CrossRef]
- Opie, L.H.; Kowolik, H. The discovery of captopril: From large animals to small molecules. Cardiovasc. Res. 1995, 30, 18–25. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, L.; Liu, Y.; Li, W.; Cai, J.; Zhang, Y. Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by supppressing proliferation, angiogenesis, and NF-KB/STAT3-regulated proteins. Cell Death Dis. 2020, 11, 477490. [Google Scholar]
- Gayathri, E.; Punnangai, K.; Chellathai, D. Evaluation of anticancer activity of olmesartan and ramipril on A549 cell line. Biomed. Pharmacol. J. 2018, 11, 1351–1357. [Google Scholar] [CrossRef]
- De Groot-Besseling, R.R.J.; Ruers, T.J.M.; van Kraats, A.A.; Poelen, G.J.M.; Ruiter, D.J.; de Waal, R.M.W.; Westphal, J.R. Anti-tumor activity of a combination of plasminogen activator and captopril in a human melanoma xenograft model. Intern. J. Cancer. 2006, 6, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Shebl, R.I. Anti-cancer potential of captopril and botulinum toxin type A and associated p53 gene apototic stimulating activity. Indian J. Pharmac. Res. 2019, 18, 1967–1977. [Google Scholar]
- Cockroft, J.R. ACE inhibition in hypertension: Focus on perindopril. Am. J. Cardiovasc. Drugs. 2007, 7, 303–317. [Google Scholar] [CrossRef]
- Ferrari, R. Angiotensin-converting enzyme inhibition in cardiovascular disease evidence with perindopril. Expert Rev. Cardiovasc. Ther. 2005, 3, 15–29. [Google Scholar] [CrossRef]
- Louis, W.J.; Conway, E.L.; Krum, H.; Workman, B.; Drummer, O.H.; Lam, W.; Philips, P.; Howes, L.G.; Jackson, B. Comparison of the pharmacokinetics and pharmacodynamics of perindopril, cilazapril and enalapril. Clin. Exp. Pharmacol. Physiol. Suppl. 1992, 19, 55–60. [Google Scholar] [CrossRef]
- Patel, S.S.; Nakka, S. Protective Effect of Perindopril on Tumor Progression and Angiogenesis in Animal Model of Breast Cancer. Anti-Cancer Agents Med. Chem. 2017, 17, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Yasumatsu, R.; Nakashima, T.; Masuda, M.; Ito, A.; Kuratomi, Y.; Nakagawa, T.; Komune, S.J. Effects of the angiotensin-I converting enzyme inhibitor perindopril on tumor growth and angiogenesis in head and neck squamous cell carcinoma cells. J. Cancer Res. Clin. Oncol. 2004, 130, 567–573. [Google Scholar] [CrossRef]
- Deshayes, F.; Nahmias, C. Angiotensin receptors: A new role in cancer? Trends Endocrinol. Metab. 2005, 16, 293–299. [Google Scholar] [CrossRef]
- Chow, L.; Rezmann, L.; Catt, K.J.; Louis, W.J.; Frauman, A.G.; Nahmias, C.; Louis, S.N. Role of the renin-angiotensin system in prostate cancer. Mol. Cell. Endocrinol. 2009, 302, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Fox, K. Contribution of perindopril to cardiology: 20 years of success. Eur. Heart J. 2007, 9, E10–E19. [Google Scholar] [CrossRef]
- Buda, V.; Minodora, A.; Adriana, L. Comparative Solid-State Stability of Perindopril Active Substance vs. Pharmaceutical Formulation. Int. J. Mol. Sci. 2017, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNicolantonio, J.J.; Lavie, C.J.; O’Keefe, J.H. Not all angiotensin-converting enzyme inhibitors are equal: Focus on ramipril and perindopril. Postgrad Med. 2013, 125, 154–168. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, H.; Chen, W.; Cui, Y.; Huang, A.; Qi, X. Perindopril improves cardiac function by enhancing the expression of SIRT3 and PGC-1α in a rat model of isoproterenol-induced cardiomyopathy. Front. Pharmacol. 2020, 11, 94. [Google Scholar] [CrossRef]
- Ancion, A.; Tridetti, J.; Trung, M.L.N.; Oury, C.; Lancellotti, P. A review of the role of bradykinin and nitric oxide in the cardioprotective action of Angiotensyn-Converting Enzyme inhibitors: Focus on perindopril. Cardiol. Ther. 2019, 8, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Prakash, K.G.; Bannur, B.M.; Madhavrao, C.; Saniya, K.; Viveka, S.; Sudha, M.J. Anti-depressant and neuroprotective effects of captopril and perindopril in paraquat model of parkinsonism. Biomed. Pharmacol. J. 2019, 12, 1715–1722. [Google Scholar] [CrossRef]
- Telejko, E. Perindopril arginine: Benefits of a new salt of the ACE inhibitor perindopril. Curr. Med. Res. Opin. 2007, 23, 953–960. [Google Scholar] [CrossRef]
- Sweetman, S.C. Martindale—The Complete Drug Reference, 37th ed.; The Pharmaceutical Press: London, UK, 2011. [Google Scholar]
- Anderson, P.J.; Critchley, J.A.; Tomlinson, B.; Resplandy, G. Comparison of the pharmacokinetics and pharmacodynamics of oral doses of perindopril in normotensive Chinese and Caucasian volunteers. Br. J. Clin. Pharmacol. 1995, 39, 361–368. [Google Scholar]
- Medenica, M.; Ivanović, D.; Masković, M.; Jancić, B.; Malenović, A. Evaluation of impurities level of perindopril tert-butylamine in tablets. J. Pharmac. Biomed. Anal. 2007, 44, 1087–1094. [Google Scholar] [CrossRef]
- European Pharmacopoeia, 10th ed.; Council of Europe Publisher: Strasbourg, France, 2020.
- Harn, Z.; Furlan, B. Process for the Preparation of Perindopril Erbumine. U.S. Patent No. 20100016614, 21 January 2010. [Google Scholar]
- Abassi, Z.; Winaver, J.; Feuerstein, G.Z. The biochemical pharmacology of renin inhibitors: Implication for translational medicine in hypertension, diabetic nephropathy and heart failure: Expectations and reality. Biochem. Pharmacol. 2009, 78, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Remko, M. Acidity, lipophilicity, solubulity, absorption, and polar surface area of some ACE inhibitors. Chem. Pap. 2007, 61, 133–141. [Google Scholar] [CrossRef]
- Kim, J.; Zhang, J.; Cha, Y.; Kolitz, S.; Funt, J.; Chong, R.E.; Barrett, S.; Kusko, R.; Zeskind, B.; Kaufman, H. Advanced bioinformatics rapidly identifies existing therapeutics for patients with coronavirus disease-2019 (COVID-19). J. Transl. Med. 2020, 18, 257. [Google Scholar] [CrossRef]
- Zisman, L.S. ACE and ACE2: A tale of two enzymes. Eur. Heart. J. 2005, 26, 32. [Google Scholar] [CrossRef]
- Harrison, C.; Acharya, K.R. ACE for all—a molecular perspective. J. Cell Commun. Signal 2014, 8, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Chang, C.; Liu, H.; Li, B.; Yan, Q.; Jiang, Z. Identification of novel angiotensyn I-converting enzyme (ACE) inhibitory peptides from wheat gluten hydrolysate by the protease of Pseudomonas aeruginosa. J. Funct. Foods 2020, 65, 103751. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Breza, M.; Madura, I.D.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. A supramolecular approach to structure-based design with a focus on synthon hierarchy in ornithine-derived ligands: Review, synthesis, experimental and in silico studies. Molecules 2020, 25, 1135. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Cole, J.C. The use of small-molecule structures to complement protein-ligand crystal structures in drug discovery. Acta Cryst. D 2017, 73, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.C.; Giangreco, I.; Groom, C.R. Using more than 801 296 small-molecule crystal structures to aid in protein structure refinement and analysis. Acta Crystallogr. D 2017, 73, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering-A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Wuest, J.D. Engineering Crystals by the Strategy of MolecularTectonics. Chem. Commun 2005, 5830–5837. [Google Scholar] [CrossRef] [PubMed]
- Corpinot, M.K.; Bucar, D.K. A practical guide to the design of molecular crystals. Cryst. Growth Design 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Wang, X.; Simard, M.; Wuest, J.D. Molecular tectonics. Supramol. Chem. 1995, 6, 171–178. [Google Scholar] [CrossRef]
- Resnati, G.; Metrangolo, P. Tectons definitions & scope. In Encyklopedia of Supramolecular Chemistry; Atwood, J.L., Steed, J.W., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2004; Volume 1, pp. 628–635. [Google Scholar]
- Rajalakshmi, P.; Srinivasan, N.; Krishnakumar, R.V.; Razak, I.A.; Rosli, M.M. Supramolecular architectures of N-acetyl-L-proline monohydrate and N-benzyl-L-proline. Acta Cryst. C 2013, 69, 1390–1396. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Madura, I.D.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Synthesis, experimental and in silico studies of N fluorenylmethoxycarbonyl-O-tert-butyl-N-methyltyrosine, coupled with CSD data: A survey of interactions in the crystal structures of Fmoc–amino acids. Acta Cryst. C 2020, 76, 328–345. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Wojciechowski, J.; Madura, I.; Kaczmarek, K.; Zabrocki, J.; Zimecki, M.; Wolf, W.M. Cyclic tetrapeptides as promising scaffold for innovative therapeutic agents: Synthesis, crystallographic, biological and in silico studies. Z. Kristallogr. Suppl. 2020, 40. [Google Scholar]
- Bojarska, J.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Amino Acids: Molecules of life. Int. J. Nutr. Sci. 2019, 4, 1035–1037. [Google Scholar]
- Bojarska, J.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Supramolecular synthons as related to cooperativity in biocomplexes: Towards design and development of oligopeptide-based modern drugs and cosmeceuticals. Nov. Approaches Drug Des. Dev. 2019, 129, 1–27. [Google Scholar]
- Bojarska, J.; Remko, M.; Wojciechowski, J.M.; Madura, I.D.; Olczak, A.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Supramolecular synthon polymorphism in modified amino acids. Structural, conformational and energy landscapes of N-benzoyl-2′-hydroxy-3-methylisovaline. J. Mol. Struct. 2019, 1190, 11–22. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. New synthons in supramolecular chemistry of short biologically active peptides. Acta Crystallog. A 2019, 75, e588. [Google Scholar] [CrossRef]
- Bojarska, J.; Kaczmarek, K.; Zabrocki, J.; Wolf, W.M. Supramolecular Chemistry of Modified Amino Acids and Short Peptides. In Advances in Organic Synthesis; Rahman, A., Ed.; Bentham Science Publishers Ltd.: Sharjah, UAE, 2018; Volume 11, pp. 43–107. [Google Scholar]
- Spackman, P.R.; Yu, L.J.; Morton, C.J.; Parker, M.W.; Bond, C.S.; Spackman, M.A.; Jayatilaka, D.; Thomas, S.P. Bridging crystal engineering and drug discovery by utilizing intermolecular interactions and molecular shapes in crystals. Angew. Chem. Int. Ed. 2019, 58, 16780–16784. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myng, S.; Pink, M.; Baik, M.H.; Clemmer, D.E. D,L-proline. Acta Crystallogr. C 2005, 61, o506–o508. [Google Scholar] [CrossRef]
- Grant, N.; Ward, M.F.; Jaspars, M.; Harrison, W.T.A. (R)-1,3-thiazolidin-3-ium-4-carboxylate. Acta Crystallogr. E 2001, 57, o697–o699. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J.; Kallen, R.G.; Voet, D. The X-ray structure of thiazolidine-4-carboxylic acid. Arch. Biochem. Biophys. 1973, 157, 426–430. [Google Scholar] [CrossRef]
- Flaig, R.; Koritsanszky, T.; Dittrich, B.; Wagner, A.; Luger, P. Intra- and Intermolecular Topological Properties of Amino Acids: A Comparative Study of Experimental and Theoretical Results. J. Am. Chem. Soc. 2002, 124, 3407–3417. [Google Scholar] [CrossRef]
- Janczak, J.; Luger, P. L-proline monohydrate at 100 K. Acta Crystallogr. C 1997, 53, 1954–1956. [Google Scholar] [CrossRef]
- Klussmann, M.; White, A.J.; Armstrong, A.; Blackmond, D.G. Rationalization and prediction of solution enantiomeric excess in ternary phase systems. Angew. Chem. Int. 2006, 45, 7985–7989. [Google Scholar] [CrossRef]
- Koetzle, T.F.; Lehmann, M.S.; Hamilton, W.C. Precision neutron diffraction strucrure determination of protein and nucleic acid components. The crystal and molecular structure of 4-hydroxy-L-proline. Acta Crystallogr. B 1973, 29, 231–236. [Google Scholar] [CrossRef]
- Schollmeyer, D. CSD Communication (Private communication). 2016. Available online: https://www.ccdc.cam.ac.uk (accessed on 23 October 2020).
- Bojarska, J.; Maniukiewicz, W.; Sieron, L.; Kopczacki, P.; Walczynski, K.; Remko, M. Perindoprilat monohydrate. Acta Crystallogr. C 2012, 68, o443–o446. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Maniukiewicz, W.; Sieron, L.; Fruzinski, A.; Kopczacki, P.; Walczynski, K.; Remko, M. Novel pseudopolymorph of the active metabolite of perindopril. Acta Crystallogr. C 2012, 68, o341–o343. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Maniukiewicz, W.; Sieron, L.; Remko, M. An orthorhombic polymorph of a cyclization product of perindopril. Acta Crystallogr. C 2013, 69, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Bojarska, J.; Maniukiewicz, W.; Główka, M.L.; Sieroń, L.; Remko, M. Crystal structure of perindopril cyclization product. J. Chil. Chem. Soc. 2013, 58, 1530–1532. [Google Scholar] [CrossRef] [Green Version]
- Bojarska, J.; Maniukiewicz, W.; Fruziński, L.; Sieron, L.; Remko, M. Captopril and its dimer captopril disulfide: Comparative structural and conformational studies. Acta Cryst. C 2015, 71, 199–203. [Google Scholar] [CrossRef]
- Remko, M.; Bojarska, J.; Jezko, J.; Sieron, L.; Olczak, A.; Maniukiewicz, W. Crystal and molecular structure of perindopril erbumine salt. J. Mol. Struct. 2011, 997, 103–109. [Google Scholar] [CrossRef]
- Remko, M.; Bojarska, J.; Jezko, L.; Olczak, A.; Maniukiewicz, W. Molecular structure of antihypertensive drug perindopril, its active metabolite perindoprilat and impurity F. J. Mol. Struct. 2013, 1036, 292–297. [Google Scholar] [CrossRef]
- Remko, M.; Bojarska, J.; Remkova, A.; Maniukiewicz, W. Molecular structure and acidity of captopril, zofenopril and their metabolites captopril disulfide and zofenoprilat. Comput. Theor. Chem. 2015, 1062, 50–55. [Google Scholar] [CrossRef]
- Wyvratt, M.J.; Tristram, E.W.; Ikeler, T.J.; Lohr, N.S.; Joshua, H.; Springer, J.P.; Arison, B.H.; Patchett, A.A. Reductive amination of ethyl 2-oxo-4-phenylbutanoate with L-alanyl-L-proline. Synthesis of enalapril maleate. J. Org. Chem. 1984, 49, 2816–2819. [Google Scholar] [CrossRef]
- Precigoux, G.; Geoffre, S.; Leroy, F. N-1-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-L-prolinium-hydrogen maleate (1/1), enalapril (MK-421). Acta Crystallogr. C 1986, 42, 1022–1024. [Google Scholar] [CrossRef]
- Fuji, K.; Uekusa, H.; Itoda, N.; Yonemochi, E.; Terada, K. Mechanism of Dehydration-Hydration Processes of Lisinopril Dihydrate Investigated by ab Initio Powder X-ray Diffraction Analysis. Cryst. Growth Des. 2012, 12, 6165–6172. [Google Scholar] [CrossRef]
- Sorrenti, M.; Catenacci, L.; Cruishank, D.L.; Caira, M.R. Lisinopril dihydrate. J. Pharm. Sci. 2013, 102, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G.; Lust, D.A.; Colapret, K.A.; Simpson, J.H.; Malley, M.F.; Gougoutas, J.Z. Sulfonation with Inversion by Mitsunobu Reaction: An Improvement on the Original Conditions. J. Org. Chem. 1996, 61, 7955–7958. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Chattopadhyay, B.; Chakraborty, S.; Roy, B.N.; Singh, G.P.; Godbole, H.M.; Rananaware, U.B.; Mukherjee, A.K. Tris(hydroxymethyl) aminomethane salt of ramipril: Synthesis, structural characterization from X-ray powder diffraction and stability studies. J. Pharm. Biomed. Anal. 2012, 70, 280. [Google Scholar] [CrossRef] [PubMed]
- Nagel, N.; Schweitzer, H.; Heyse, W.; Muller, B.; Berchtold, H. Ramipril. Acta Crystallogr. E 2001, 57, o463–o465. [Google Scholar] [CrossRef]
- Paulus, E.F.; Henning, R.; Urbach, H. Absolute configuration and structure of ethyl 2-(3-methyl-1-4-dioxoperhydrocyclopenta[4,5]pyrrolo[1,2]pyrazin-2-yl)4-phnylbutyrate. Acta Crystallogr. C 1987, 43, 938–941. [Google Scholar] [CrossRef]
- Reid, J.W.; Kaduk, J.A.; Vickers, M. The crystal structure of trandolapril. Powder Diffr. 2016, 31, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wahle, M.C.; Stowell, J.G.; Byrn, S.R. Spirapril Hydrochloride Hydrate. Acta Crystallogr. C 1997, 53, 1917–1919. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. A general definition of ring puckering coordinates. J.Amer. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Schmidt, M.; Brader, J.M. Power functional theory for Brownian dynamics. J. Chem. Phys. 2013, 138, 214101–214102. [Google Scholar] [CrossRef] [Green Version]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCr J. 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Akif, M.; Georgiadis, D.; Mahajan, A.; Dive, V.; Sturrock, E.D.; Isaac, R.E.; Acharya, K.R. Crystal structure of AnCE-perindopril complex. J. Mol. Biol. 2010, 400, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.S.; Cozier, G.E.; Harrison, C.; Isaac, R.E.; Acharya, K.R. Crystal structures of angiotensin-converting enzyme from Anopheles gambiae in its native form and with a bound inhibitor. Biochem. J. 2019, 476, 3505–3520. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Desiraju, G.R. Long-range synthon Aufbau modules (LSAM) in crystal structures: Systematic changes in C6H6−nFn (0 ≤ n ≤ 6) fluorobenzenes. Cryst. Eng. Commun. 2010, 12, 817–833. [Google Scholar] [CrossRef]
- Liu, K.T.; Chen, C.H. Determination of impurities in pharmaceuticals: Why and how? In Quality Management and Quality Control—New Trends and Developments; IntechOpen: London, UK, 2019. [Google Scholar]
- Rayavarapu, S.; Braithwaite, E.; Dorsam, R.; Osterhout, J.; Furlong, L.A.; Shetty, D. Comparative risk assessment of formulation changes in generic drug products: A pharmacology/toxicology perspective. Toxicol. Sci. 2015, 146, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, W.C.; Kenyon, M.O.; Cheung, J.R.; Dugger, R.W.; Dobo, K.L. Resolution of contradiction between in silico predictions and Ames test results for four pharmaceutically relevant impurities. Regul. Toxicol. Pharmacol. 2017, 91, 68–76. [Google Scholar] [CrossRef]
- Pikul, P.; Jamrogiewicz, M.; Nowakowska, J.; Hewelt-Belka, W.; Ciura, K. Forced degradation studies of ivabradine and in silico toxicologynpredictions for its new designated impurities. Front. Pharmacol. 2016, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Kragelj, L.N.; Toplak, C.R.; Jurca, S.; Doljak, B. Theoretical purge factor determination as a control strategy for potential mutagenic impurities in the synthesis of drug substances. Acta Chim. Slov. 2017, 64, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Salunkhe, M.N.; Gite, S.D.; Kachave, R.N. Recent trends in impurity profiling and forced degradation of antihypertensive drugs. J. Liquid Chromatogr. 2017, 40, 813–831. [Google Scholar] [CrossRef]
- Remko, M. Molecular structure and stability of perindopril erbumine and perindopril L-arginine complexes. Eur. J. Med. Chem. 2009, 44, 101–108. [Google Scholar] [CrossRef]
- Szabo, Z.I.; Reti, Z.Z.; Gagyi, L.; Kis, E.L.; Sipos, E. Simultaneous quantification of related substances of perindopril tert-butylamine using a novel stability indicating liquid chromatographic method. J. Chromatogr. Sci. 2015, 53, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42730. [Google Scholar] [CrossRef] [Green Version]
- Qidwai, T. QSAR modeling, docking and ADMET studies for explorationof potential anti-malarial compounds against Plasmodium falciparum. Silico Pharmacol. 2017, 5, 6–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonteh, P.; Elkhadir, A.; Omondi, B.; Guzei, I.; Darkwa, J.; Meyer, D. Impedance technology reveals correlations between cytotoxicity andlipophilicity of mono and bimetallic phosphine complexes. Biometals 2015, 28, 653–667. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.v.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Klamt, A.; Schűman, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, E.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural-population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural Localized Molecular Orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ugliengo, P. MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, Torino. 2006. Available online: www.moldraw.unito.it (accessed on 23 October 2020).
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Commun. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Mackenzie, M.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies, Energy Frameworks, Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCr J. 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Commun. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J.; Jayatilaka, D. Electrostatic potentials mapped on Hirshfeld surfaces provides direct insight into intermolecular interactions in crystals. Cryst. Eng. Commun. 2008, 10, 377–388. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W. ADMET-score- a comprehensive scoring function for evaluation of chemical drug-likeness. Med. Chem. Commun. 2019, 10, 148–157. [Google Scholar] [CrossRef]
- Daina, A.; Blatter, M.C.; Gerritsen, V.B.; Palagi, P.M.; Marek, D.; Xenarios, I.; Schwede, T.; Michielin, O.; Zoete, V. Drug design workshop: A web-based educational tool to introduce computer-aided drug design to the general public. J. Chem. Educ. 2017, 94, 335–344. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
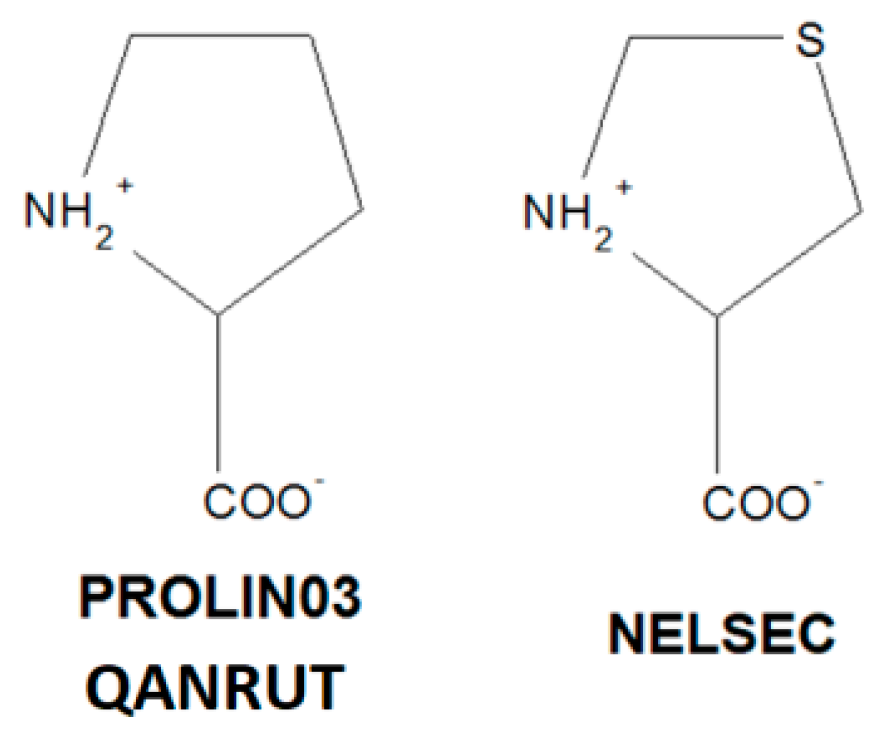{kind=link}
| I | Conformation | II | Conformation | III | Conformation |
|---|---|---|---|---|---|
 |  |  | |||
| BECWIR | trans | IVEGIA | trans | NELSEC | trans |
| FEFKEI | trans | UZOVAH03 | trans | PROLIN03 | trans |
| MCPRL01 | trans | DIVHOF01 | trans | QANRUT | trans |
| CIYNIH | trans | EDALEC | trans | ||
| IQISAE | cis | FIFGEG | trans | ||
| QOQWAU | cis | GERXAE | trans | ||
| RUWBAM | trans | GERXEI | trans | ||
| YOZTIS | trans | GERWUX01 | trans |
| Interactions | IVEGIA | UZOWAH03 | ||
|---|---|---|---|---|
| Surface (%) | H | O | ||
| 85.4 | 87.9 | 14.2 | 11.2 | |
| Major contacts | H…H | O…H | ||
| Proportion (%) | 73.8 | 76.6 | 23.2 | 22.6 |
| ER | 1 | 1.0 | 0.96 | 1.14 |
| Interactions | BECWIR | FEFKEI | ||||||
|---|---|---|---|---|---|---|---|---|
| Surface (%) | H | O | C | S | ||||
| 61.95 | 81.4 | 18 | 18.05 | 0.45 | 0.45 | 0.7 | - | |
| Major contacts | H…H | O…H | C…H | S…H | ||||
| Proportion (%) | 60.8 | 62.9 | 36 | 36.1 | 0.9 | 0.9 | 1.4 | - |
| ER | 1.58 | 0.95 | 1.61 | 1.23 | 1.61 | 1.23 | 1.61 | - |
| Interactions | BILNAN | BILNAN01 | ||||
|---|---|---|---|---|---|---|
| Surface (%) | H | O | N | |||
| 86.3 | 88.25 | 11.35 | 10.35 | 0.45 | ||
| Major contacts | H…H | O…H | N…H | |||
| Proportion (%) | 74.5 | 77.9 | 22.7 | 20.7 | 0.9 | |
| ER | 1.0 | 1.0 | 1.16 | 1.1 | 1.15 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojarska, J.; Remko, M.; Breza, M.; Madura, I.; Fruziński, A.; Wolf, W.M. A Proline-Based Tectons and Supramolecular Synthons for Drug Design 2.0: A Case Study of ACEI. Pharmaceuticals 2020, 13, 338. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110338
Bojarska J, Remko M, Breza M, Madura I, Fruziński A, Wolf WM. A Proline-Based Tectons and Supramolecular Synthons for Drug Design 2.0: A Case Study of ACEI. Pharmaceuticals. 2020; 13(11):338. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110338
Chicago/Turabian StyleBojarska, Joanna, Milan Remko, Martin Breza, Izabela Madura, Andrzej Fruziński, and Wojciech M. Wolf. 2020. "A Proline-Based Tectons and Supramolecular Synthons for Drug Design 2.0: A Case Study of ACEI" Pharmaceuticals 13, no. 11: 338. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110338