Synthesis of New 1, 3, 4-Oxadiazole-Incorporated 1, 2, 3-Triazole Moieties as Potential Anticancer Agents Targeting Thymidylate Synthase and Their Docking Studies

 , , ,
, , ,
Abstract
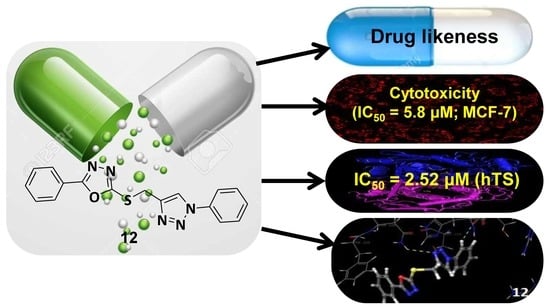
:
1. Introduction
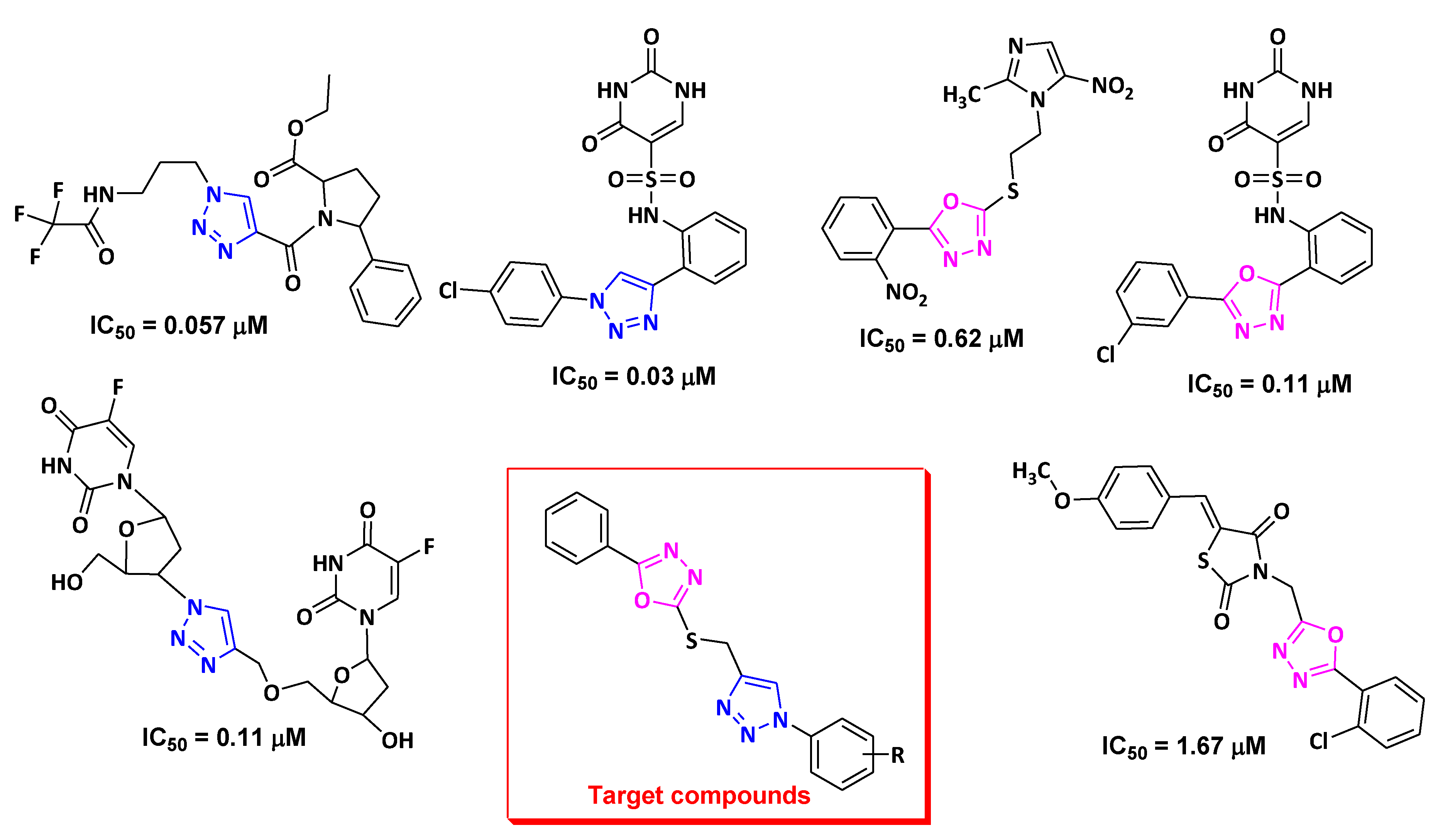
2. Results and Discussion
2.1. Chemistry
2.2. Pharmacokinetics Studies/ADME Predictions
2.3. Biological Studies
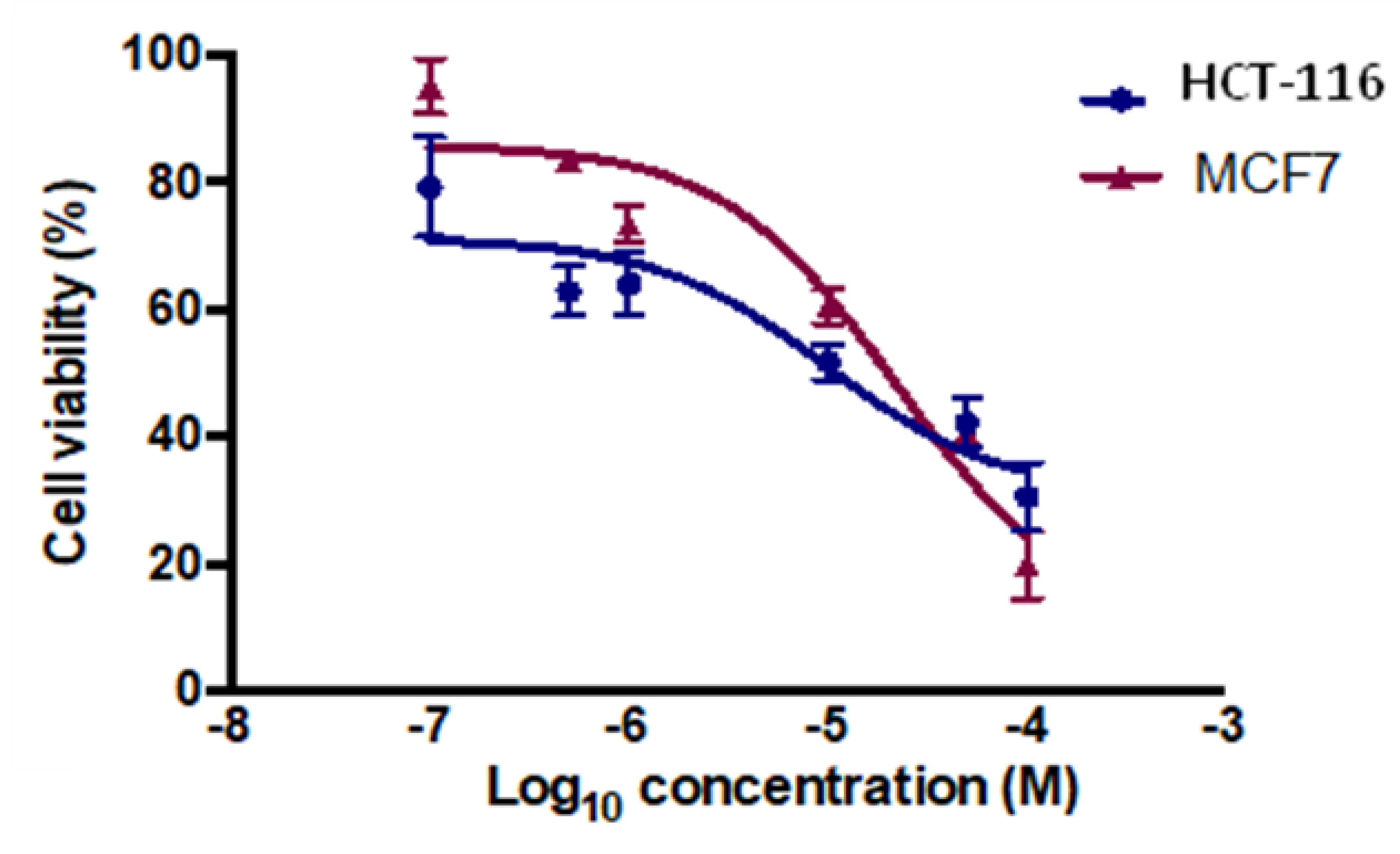
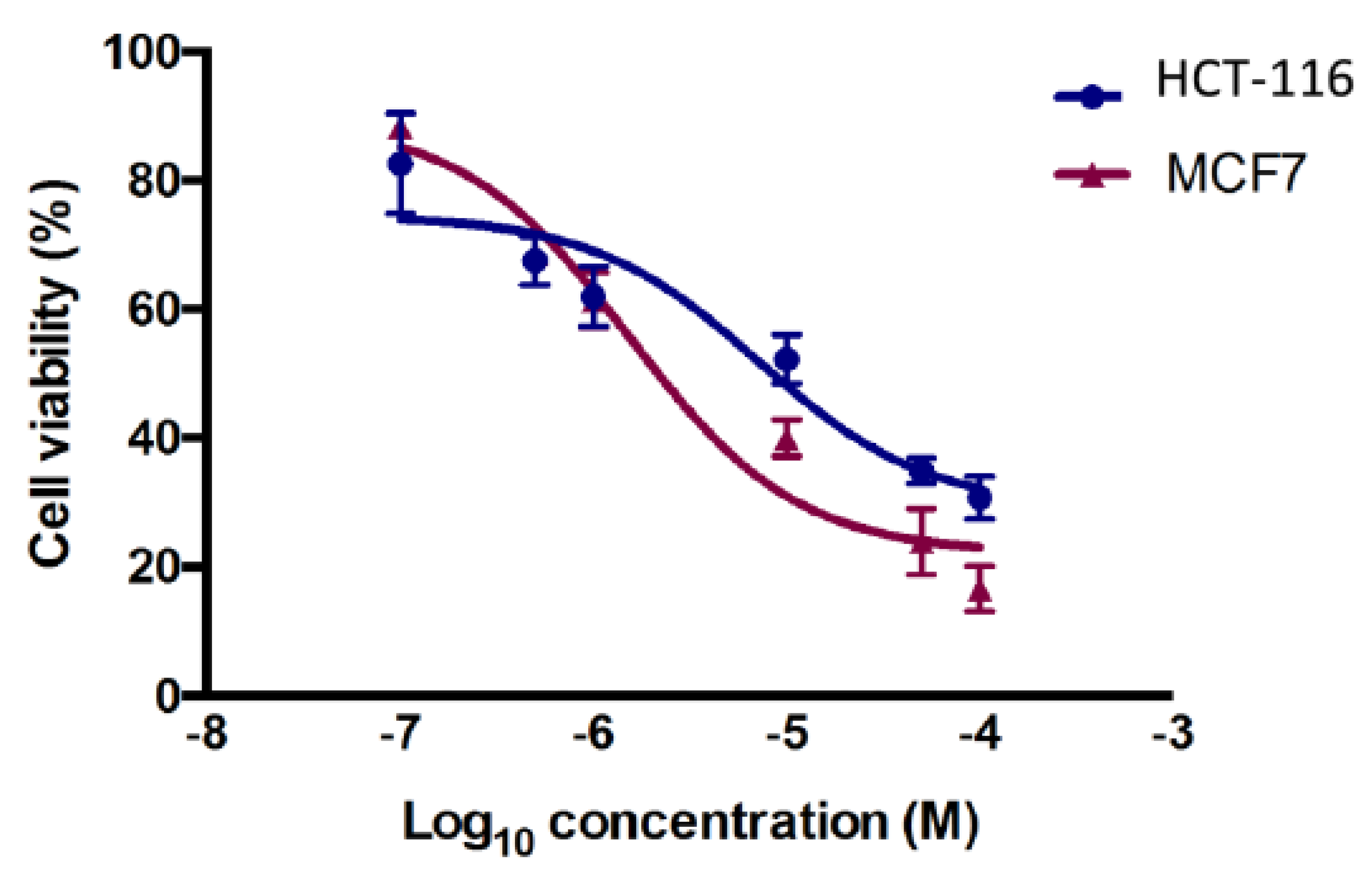
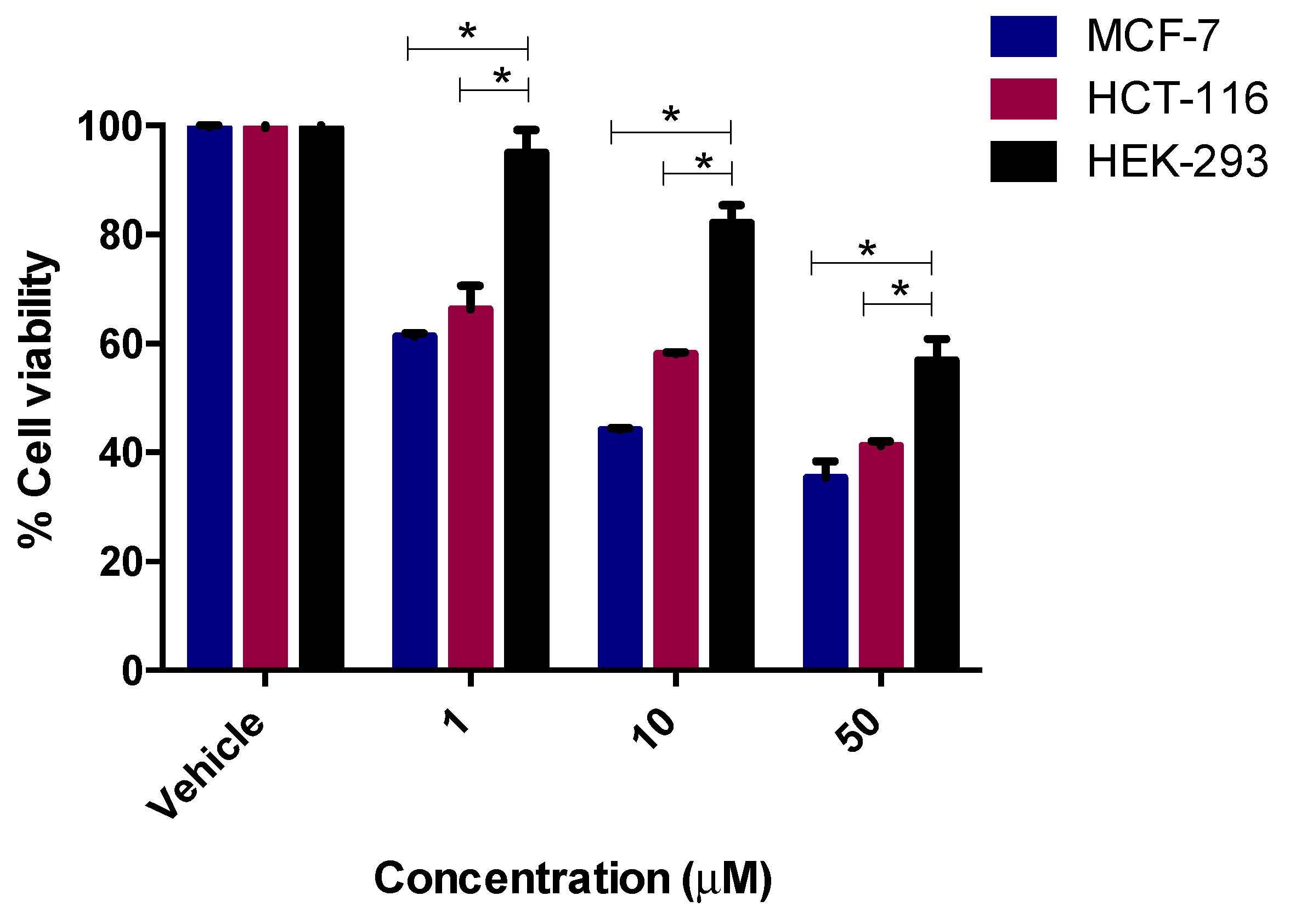
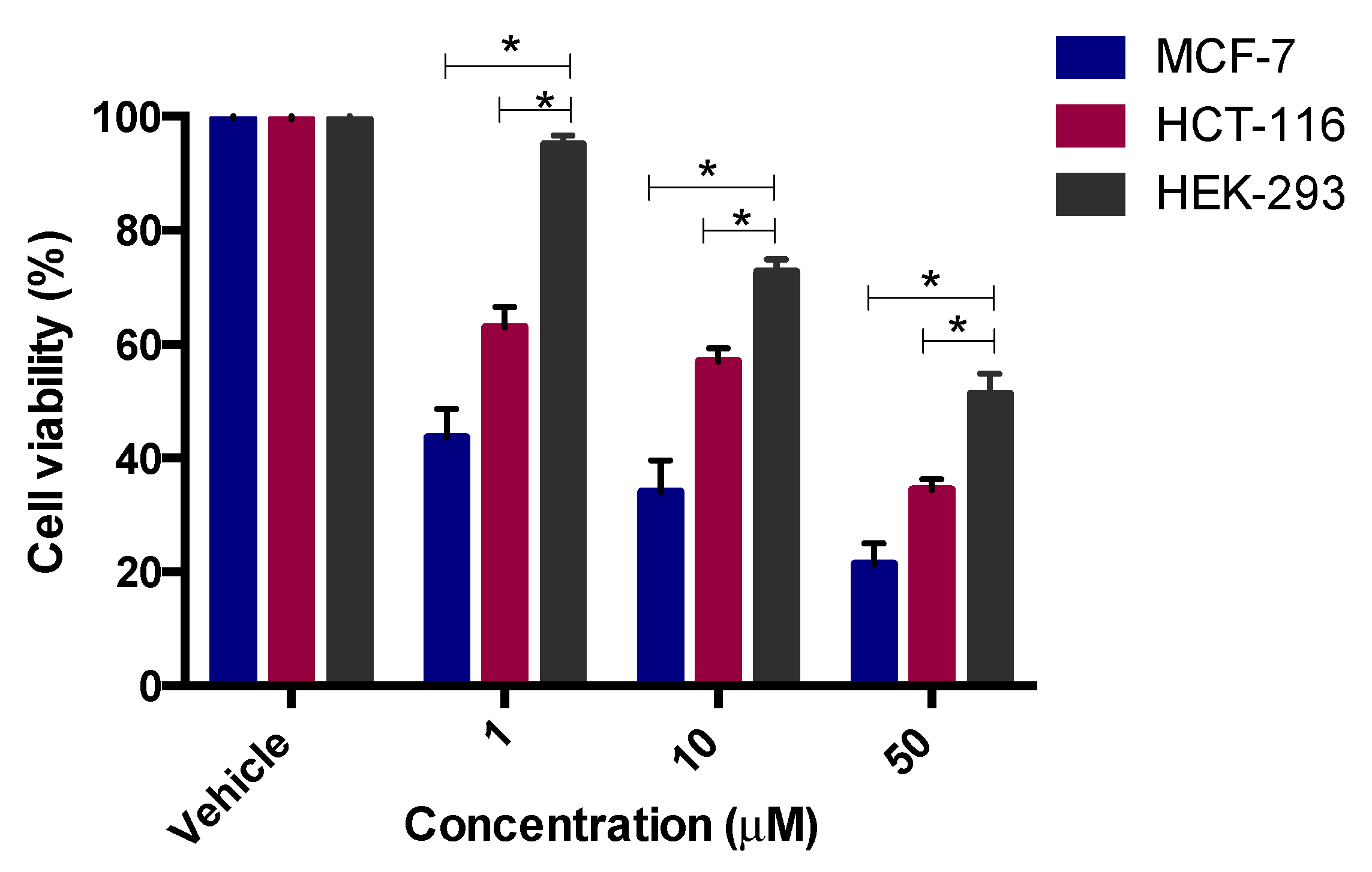
2.3.1. Cytotoxicity Assay
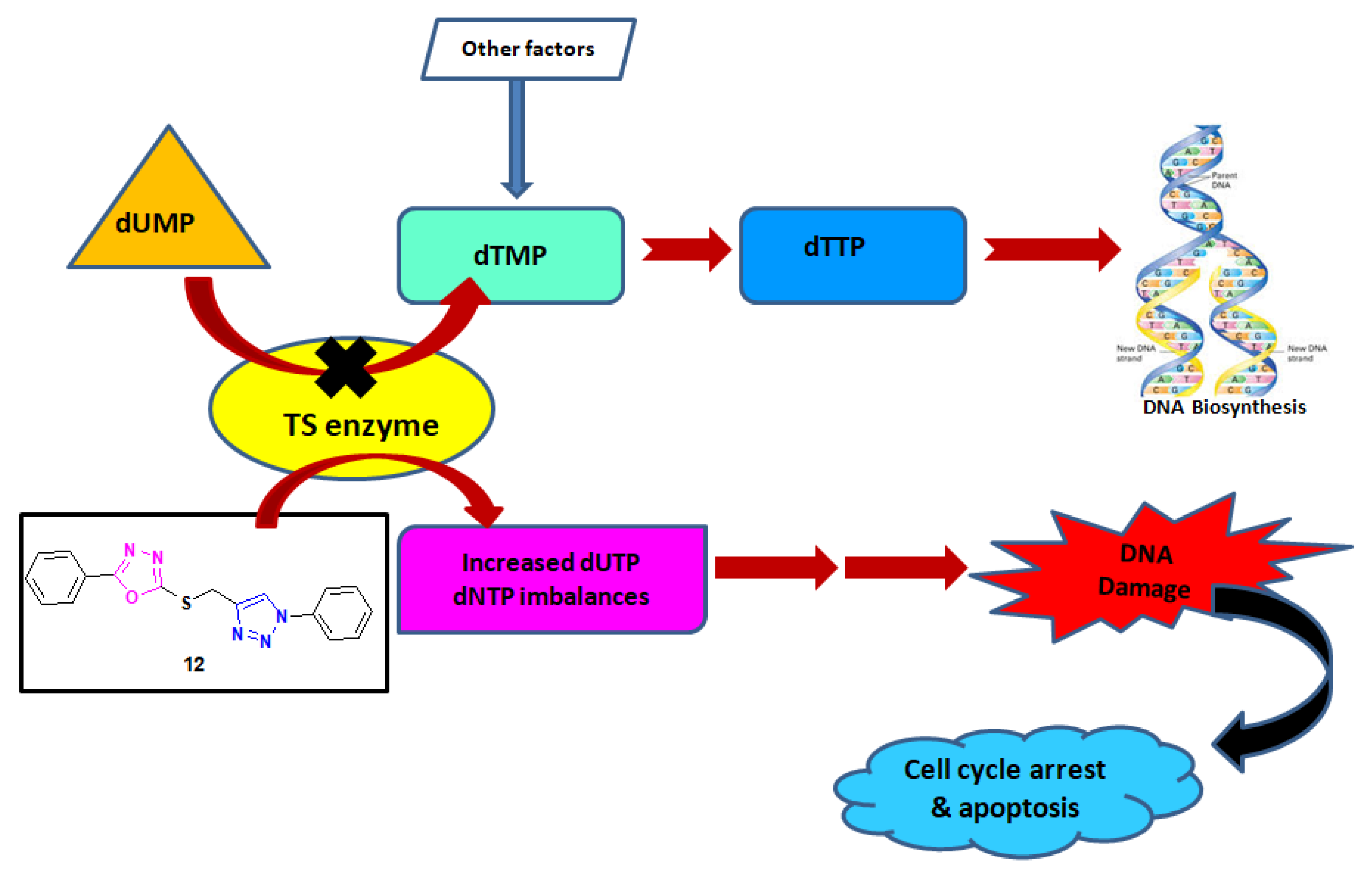
2.3.2. In Vitro Thymidylate Synthase Activity
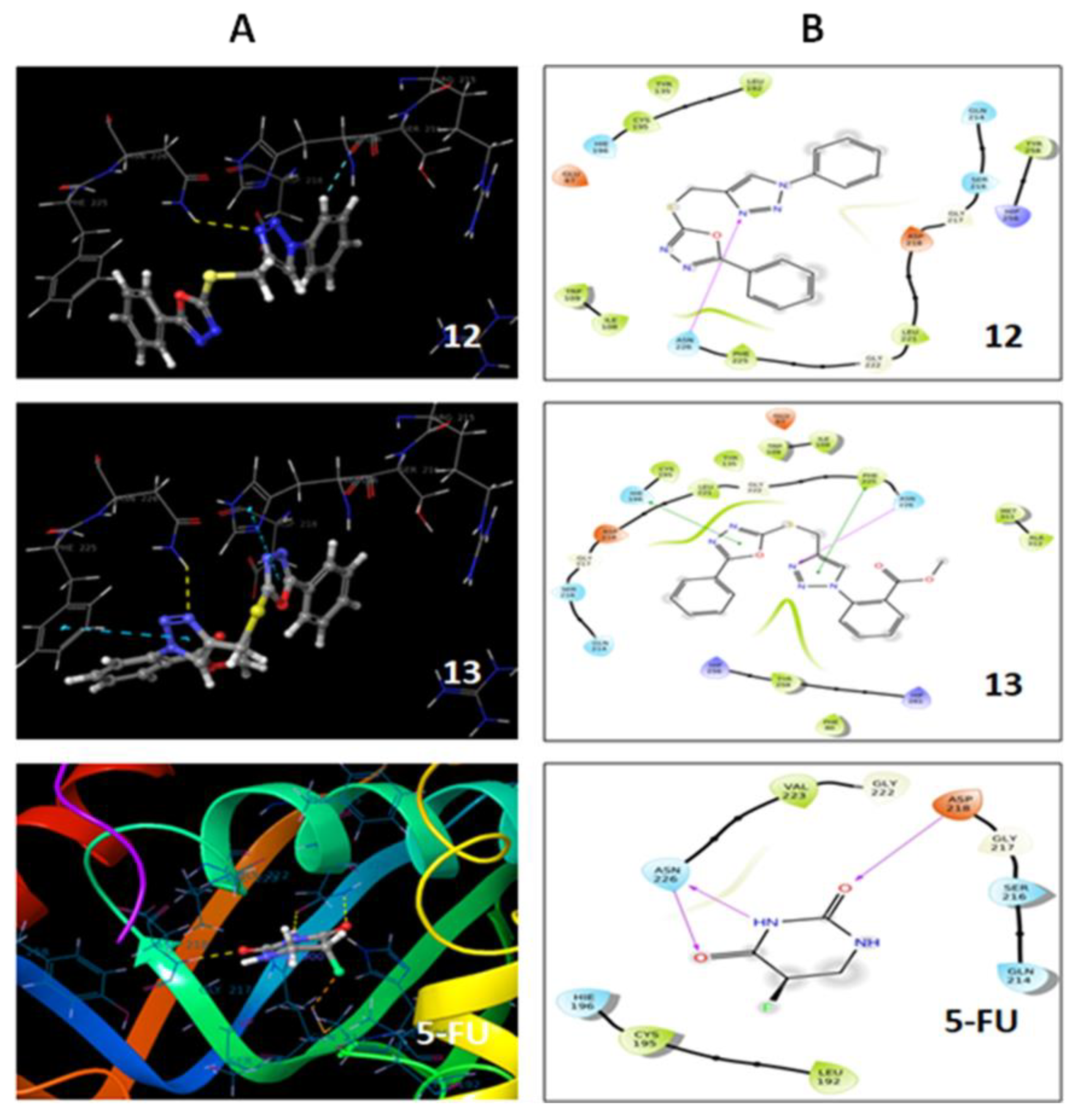
2.4. Molecular Docking
3. Experimental
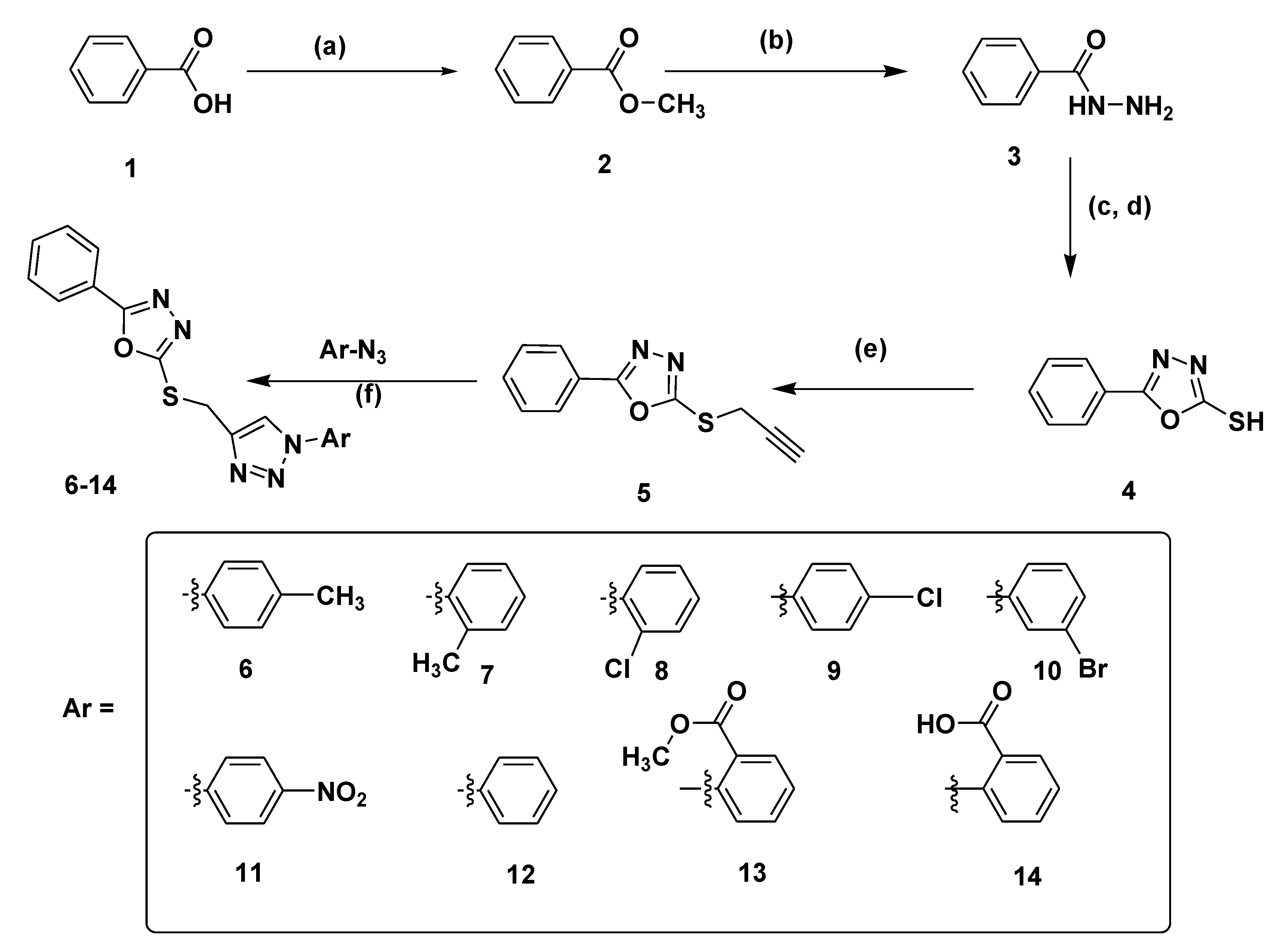
3.1. Chemistry
3.2. General Procedure for the Synthesis of 1,3,4-Oxadiazole Linked 1,2,3-Triazole Hybrids (6–14)
3.2.1. 4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)-1-p-tolyl–1H-1,2,3-triazole (6)
3.2.2. 4-((5-phenyl–1,3,4-oxadiazol-2-ylthio)methyl)–1-o-tolyl-1H–1,2,3-triazole (7)
3.2.3. 4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)–1-(2-chlorophenyl)-1H–1,2,3-triazole (8)
3.2.4. 4-((5-phenyl–1,3,4-oxadiazol-2-ylthio)methyl)–1-(4-chlorophenyl)-1H–1,2,3-triazole (9)
3.2.5. 4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)-1-(3-bromophenyl)-1H–1,2,3-triazole (10)
3.2.6. 4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)–1-(4-nitrophenyl)-1H–1,2,3-triazole (11)
3.2.7. 4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)-1-phenyl-1H–1,2,3-triazole (12)
3.2.8. Methyl 2-(4-((5-phenyl–1,3,4-oxadiazol-2-ylthio)methyl)-1H–1,2,3-triazol–1-yl)benzoate (13)
3.2.9. 2-(4-((5-phenyl–1,3,4-oxadiazol–2-ylthio)methyl)-1H–1,2,3-triazol–1-yl)benzoic acid (14)
3.3. Anticancer Activity
3.3.1. Cell Lines and Culture Medium
3.3.2. Cytotoxicity Assay
3.4. In Vitro Thymidylate Synthase Enzyme Assay
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanism of Cancer drug resistance. A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 769–1792. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Liang, J.W.; Mohamed, K.O.; Zhang, T.J.; Lu, G.Q.; Meng, F.H. Design, synthesis and biological evaluation of N-phenyl-(2,4- dihydroxypyrimidine-5-sulfonamido)benzoyl hydrazide derivatives as thymidylate synthase (TS) inhibitors and as potential antitumor drugs. Eur. J. Med. Chem. 2018, 154, 267–279. [Google Scholar] [CrossRef]
- Carreras, C.W.; Santi, D.V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef]
- Rode, W.; Les, A. Molecular mechanism of thymidylate synthase-catalyzed reaction and interaction of the enzyme with 2- and/or 4-substituted analogues of dUMP and 5-fluoro-dUMP. Acta Biochim. Pol. 1996, 43, 133–142. [Google Scholar] [CrossRef]
- Hardy, L.W. Structural aspects of the inhibition and catalytic mechanism of thymidylate synthase. Acta Biochim. Pol. 1995, 42, 367–380. [Google Scholar] [CrossRef]
- Kumar, V.P.; Cisneros, J.A.; Frey, K.M.; Castellanos-Gonzalez, A.; Wang, Y.; Gangjee, A.; White, A.C.; Jorgensen, W.L.; Anderson, K.S. Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase-dihydrofolate reductase. Bioorg. Med. Chem. Lett. 2014, 24, 4158–4161. [Google Scholar] [CrossRef] [Green Version]
- Catalano, A.; Luciani, R.; Carocci, A.; Cortesi, D.; Pozzi, C.; Borsari, C.; Ferrari, S.; Mangani, S. X-ray crystal structures of Enterococcus faecalis thymidylate synthase with folate binding site inhibitors. Eur. J. Med. Chem. 2016, 123, 649–664. [Google Scholar] [CrossRef] [Green Version]
- Houghton, J.A.; Tillman, D.M.; Harwood, F.G. Ratio of 2′-deoxyadenosine-5′-triphosphate/thymidine-5′-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin. Cancer Res. 1995, 1, 723–730. [Google Scholar] [PubMed]
- Aherne, G.W.; Hardcastle, A.; Raynaud, F.; Jackman, A.L. Immunoreactive dUMP and TTP pools as an index of thymidylate synthase inhibition; effect of tomudex (ZD1694) and a non polyglutamated quinazoline antifolate (CB30900) in L1210 mouse leukaemia cells. Biochem. Pharmacol. 1996, 51, 1293–1301. [Google Scholar] [CrossRef]
- Alvarez, P.; Marchal, J.A.; Boulaiz, H.; Carrillo, E.; Velez, C.; Rodríguez-Serrano, F.; Melguizo, C.; Prados, J.; Madeddu, R.; Aranega, A. 5-Fluorouracil derivatives: A patent review. Expert Opin. Ther. Pat. 2015, 22, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; McHenry, C.S.; Sommer, H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 1974, 13, 471–481. [Google Scholar] [CrossRef]
- Pinheiro, S.; Pessoa, J.C.; Pinheiro, E.M.C.; Muri, E.M.F.; Greco, S.J. 2H-1,2,3-Triazole-chalcones as novel cytotoxic agents against prostate cancer. Bioorg. Med. Chem. Lett. 2020, 30, 127457–127463. [Google Scholar] [CrossRef]
- Yan, L.; Yin, Z.; Niu, L.; Shao, J.; Chen, H.; Li, X. Synthesis of pentacyclic iminosugars with constrained butterfly-like conformation and their HIV-RT inhibitory activity. Bioorg. Med. Chem. Lett. 2018, 28, 425–428. [Google Scholar] [CrossRef]
- Saeedi, M.; Mohammadi-Khanaposhtani, M.; Pourrabia, P.; Razzagi, N.; Ghadimi, R.; Imanparast, S.; Faramarzi, M.A.; Bandarian, F.; Esfahani, E.N.; Safavi, M.; et al. Design and synthesis of novel quinazolinone-1,2,3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg Chem. 2019, 83, 161–169. [Google Scholar] [CrossRef]
- Bi, F.; Ji, S.; Venter, H.; Liu, J.; Semple, S.J.; Ma, S. Substitution of terminal amide with 1H-1,2,3-triazole: Identification of unexpected class of potent antibacterial agents. Bioorg. Med. Chem. Lett. 2018, 28, 884–891. [Google Scholar] [CrossRef]
- Moussa, G.; Alaaeddine, R.; Alaeddine, L.M.; Nasra, R.; Belal, A.S.F.; Ismail, A.; El-Yazbi, A.F.; Abdel-Ghany, Y.S.; Hazza, A. Novel click modifiable thioquinazolinones as anti-inflammatory agents: Design, synthesis, biological evaluation and docking study. Eur. J. Med. Chem. 2018, 144, 635–650. [Google Scholar] [CrossRef]
- Yan, X.; Lv, Z.; Wen, J.; Zhao, S.; Xu, Z. Synthesis and in vitro evaluation of novel substituted isatin-propylene-1H-1,2,3-triazole-4- methylene-moxifloxacin hybrids for their anti-mycobacterial activities. Eur. J. Med. Chem. 2018, 143, 899–904. [Google Scholar] [CrossRef]
- Lu, G.Q.; Li, X.Y.; Mohamed, O.K.; Wang, D.; Meng, F.H. Design, synthesis and biological evaluation of novel uracil derivatives bearing 1, 2, 3-triazole moiety as thymidylate synthase (TS) inhibitors and as potential antitumor drugs. Eur. J. Med. Chem. 2019, 171, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, D.; Baranowski, D.; Ruszkowski, P.; Boryski, J. Nucleoside dimmers anlogues with a 1,2,3-triazole linkage: Conjugation of floxuridine and thymidine provides novel tools for cancer treatment. Nucleosides Nucleotides Nucleic Acids 2019, 38, 807–835. [Google Scholar] [CrossRef] [PubMed]
- Onen, F.E.; Boum, Y.; Jacquement, C.; Spanedda, M.V.; Jaber, N.; Scherman, D.; Myllykallio, H.; Herscovici, J. Design, synthesis and evaluation of potent thymidylate synthase X inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Xi, H.; Li, C.; Bian, S.; Cheng, H.; Cui, J.; Wang, N.; Wei, B.; Huang, X.; Chen, L. Endothelin-A receptor in gastric cancer and enhanced antitumor activity of trastuzumab in combination with the endothelin-A receptor antagonist ZD4054. Ann. N.Y. Acad. Sci. 2019, 1448, 30–41. [Google Scholar] [CrossRef]
- Du, Q.R.; Li, D.D.; Pi, Y.Z.; Li, J.R.; Sun, J.; Fang, F.; Zhong, W.Q.; Gong, H.B.; Zhu, H.L. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg. Med. Chem. 2013, 21, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, D.; Lu, G.; Lu, K.; Zhang, T.; Li, S.; Mohamed, K.O.; Xue, W.; Qian, X.; Meng, F. Development of a novel thymidylate synthase (TS) inhibitor capable of up-regulating P53 expression and inhibition angiogenesis in NSCLC. J. Adv. Res. 2020, 26, 95–110. [Google Scholar] [CrossRef]
- Alzhrani, Z.M.M.; Alam, M.M.; Neamatallah, T.; Nazreen, S. Design, synthesis and in vitro antiproliferative activity of new thiazolidinedione-1,3,4-oxadiazole hybrids as thymidylate synthase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1116–1123. [Google Scholar] [CrossRef]
- Jha, K.K.; Samad, A.; Kumar, Y.; Shaharyar, M.; Khosa, R.L.; Jain, J.; Kumar, V.; Singh, P. Design, synthesis and biological evaluation of 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 4963–4967. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Freener, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lamia, H.T.A.; Taghreed, Z.S.; Abeer, M.N. Design, synthesis, anticancer evaluation and docking studies of new pyrimidine derivatives as potent thymidylate synthase inhibitors. Bioorg. Chem. 2019, 9, 103159–103173. [Google Scholar]
- Ahmed, F.E.; Qasem, M.A.A.; Emad, S.I.H. Design, synthesis, molecular docking of new thiopyrimidine-5-carbonitrile derivatives and their cytotoxic activity against HepG2 cell line. J. Appl. Pharm. Sci. 2014, 4, 102–111. [Google Scholar]
- Meerloo, J.V.; Kaspers, G.J.L.; Cloos, J. Cell sensitivity assays. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [PubMed]
- Wahba, A.J.; Friedkin, M. The enzymatic synthesis of thymidylate: I. early steps in the purification of thymidylate synthetase of Escherichia coli. J. Biol. Chem. 1962, 237, 3794–3801. [Google Scholar]
- Davisson, V.J.; Sirawaraporn, W.; Santi, D.V. Expression of human thymidylate synthase in Escherichia coli. J. Biol. Chem. 1989, 264, 9145–9148. [Google Scholar]
- Nazreen, S.; Alam, M.S.; Hamid, H.; Yar, M.S.; Shafi, S.; Dhulap, A.; Alam, P.; Pasha, M.A.Q.; Bano, S.; Alam, M.M.; et al. Design, synthesis, in silico molecular docking and biological evaluation of novel oxadiazole based thiazolidine-2,4-diones bis-heterocycles as PPAR-γ agonists. Eur. J. Med. Chem. 2014, 87, 175–185. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Lipinski Parameters | nROTB e | TPSA f | %ABS g | BBB h | GI ABS i | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| MW a | HBAs b | HBDs c | LogP d | Violations | ||||||
| 6 | 349.41 | 5 | 0 | 3.54 | 0 | 5 | 94.93 | 76.24 | No | High |
| 7 | 349.41 | 5 | 0 | 3.5 | 0 | 5 | 94.93 | 76.24 | No | High |
| 8 | 369.83 | 5 | 0 | 3.33 | 0 | 5 | 94.93 | 76.24 | No | High |
| 9 | 369.83 | 5 | 0 | 3.5 | 0 | 5 | 94.93 | 76.24 | No | High |
| 10 | 414.28 | 5 | 0 | 3.62 | 0 | 5 | 94.93 | 76.24 | No | High |
| 11 | 380.38 | 7 | 0 | 2.97 | 0 | 6 | 140.75 | 60.44 | No | Low |
| 12 | 335.38 | 5 | 0 | 3.21 | 0 | 5 | 94.93 | 76.24 | No | High |
| 13 | 393.42 | 7 | 0 | 3.39 | 0 | 7 | 121.23 | 67.17 | No | High |
| 14 | 379.39 | 7 | 1 | 2.48 | 0 | 6 | 132.23 | 63.38 | No | High |
| Compound | MCF-7 b | HCT-116 c |
|---|---|---|
| 6 | 79.80 | 89.20 |
| 7 | 30.70 | 34.30 |
| 8 | 73.30 | 107.50 |
| 9 | 34.40 | 36.70 |
| 10 | 25.90 | 32.70 |
| 11 | 98.20 | 102.30 |
| 12 | 5.80 | 14.80 |
| 13 | 1.26 | 17.30 |
| 14 | 40.60 | 46.80 |
| Tam d | 5.12 | 26.41 |
| 5-FU e | 24.74 | 32.68 |
| Compounds | IC50 (µM) |
|---|---|
| 12 | 2.52 |
| 13 | 4.38 |
| PTX | 6.75 |
| Compound | Docking Score | Amino Acid Residue |
|---|---|---|
| 12 | −3.81 | ASN 226 |
| 13 | −4.25 | ASN 226, PHE 225, HIE 196 |
| 5-FU | −3.5 | ASP 218, ASN 226 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, M.M.; Almalki, A.S.; Neamatallah, T.; Ali, N.M.; Malebari, A.M.; Nazreen, S. Synthesis of New 1, 3, 4-Oxadiazole-Incorporated 1, 2, 3-Triazole Moieties as Potential Anticancer Agents Targeting Thymidylate Synthase and Their Docking Studies. Pharmaceuticals 2020, 13, 390. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110390
Alam MM, Almalki AS, Neamatallah T, Ali NM, Malebari AM, Nazreen S. Synthesis of New 1, 3, 4-Oxadiazole-Incorporated 1, 2, 3-Triazole Moieties as Potential Anticancer Agents Targeting Thymidylate Synthase and Their Docking Studies. Pharmaceuticals. 2020; 13(11):390. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110390
Chicago/Turabian StyleAlam, Mohammad Mahboob, Abdulraheem SA Almalki, Thikryat Neamatallah, Nada M. Ali, Azizah M. Malebari, and Syed Nazreen. 2020. "Synthesis of New 1, 3, 4-Oxadiazole-Incorporated 1, 2, 3-Triazole Moieties as Potential Anticancer Agents Targeting Thymidylate Synthase and Their Docking Studies" Pharmaceuticals 13, no. 11: 390. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110390