Repeated Administration of Clinical Doses of Tramadol and Tapentadol Causes Hepato- and Nephrotoxic Effects in Wistar Rats

 ,
,  ,
, 
 and
and 
Abstract
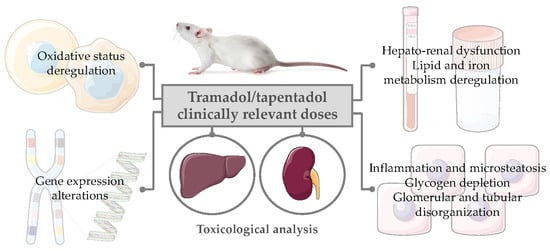
:
1. Introduction
2. Results
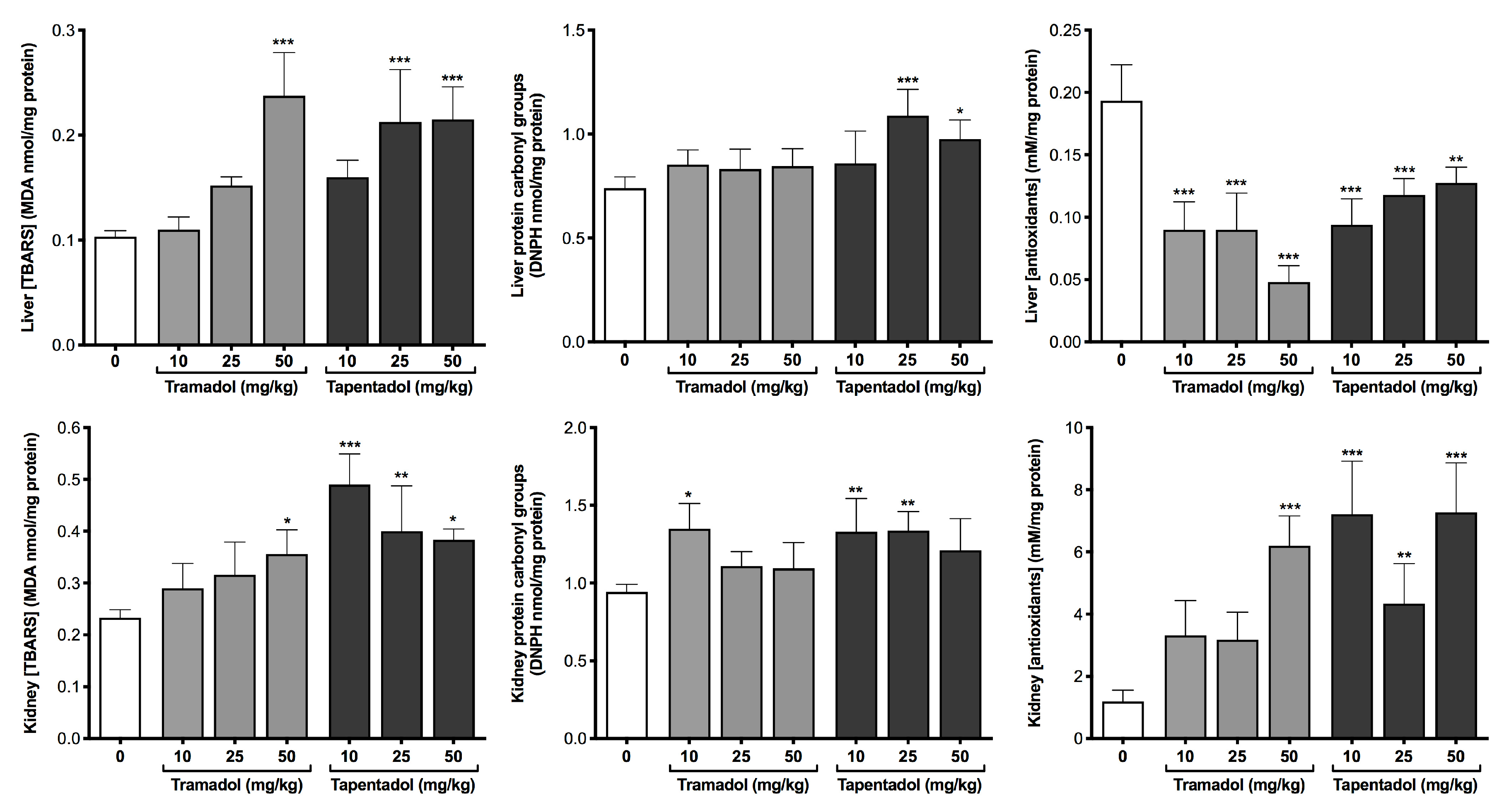
2.1. Repeated Exposure to Tramadol and Tapentadol Causes Oxidative Stress and Differentially Changes the Antioxidant Status of Liver and Kidney
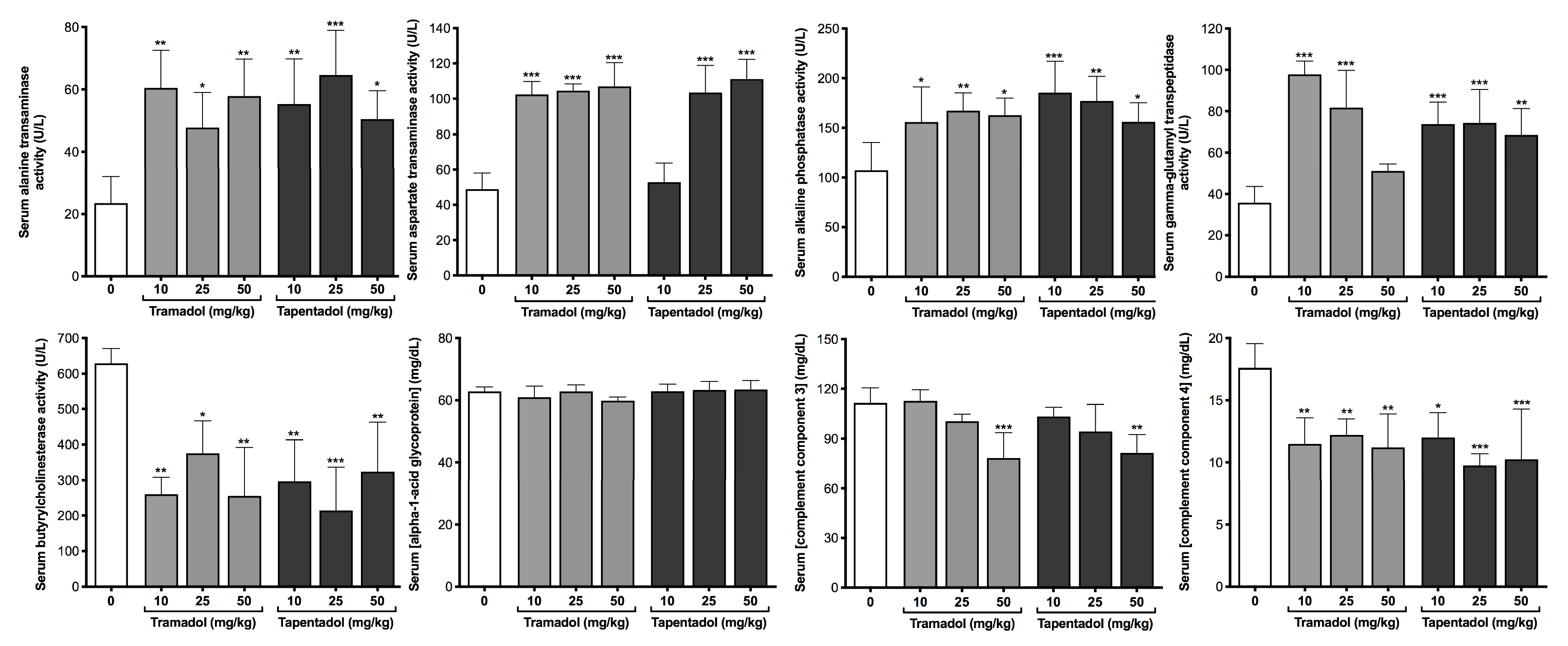
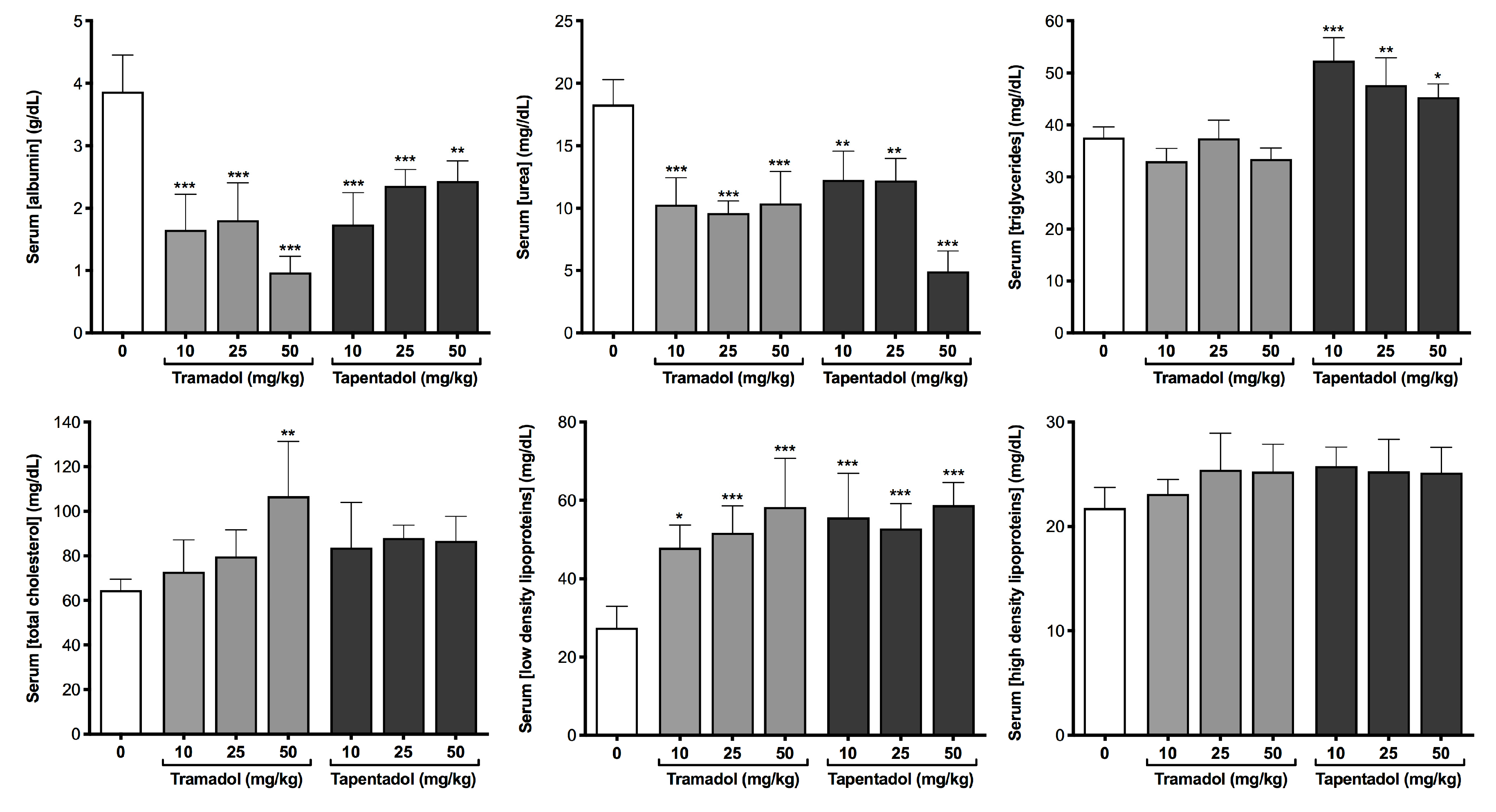
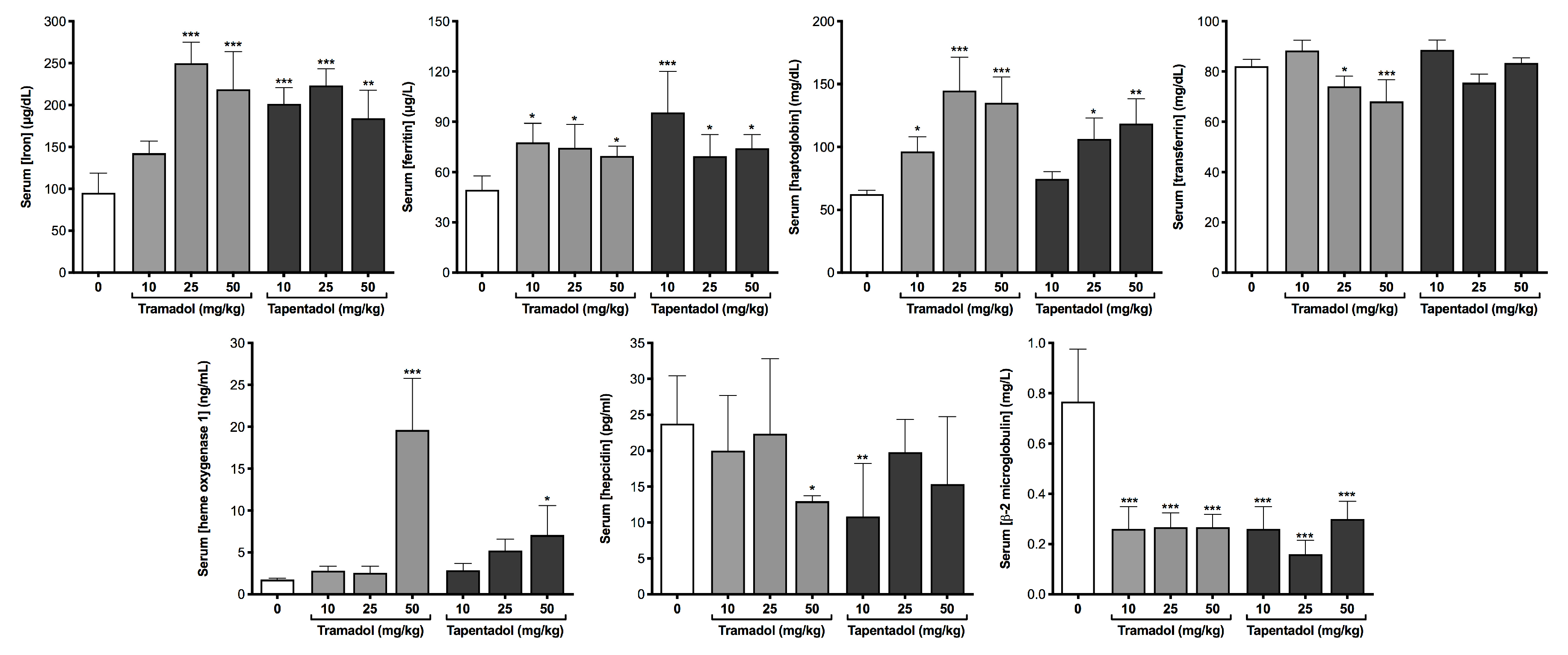
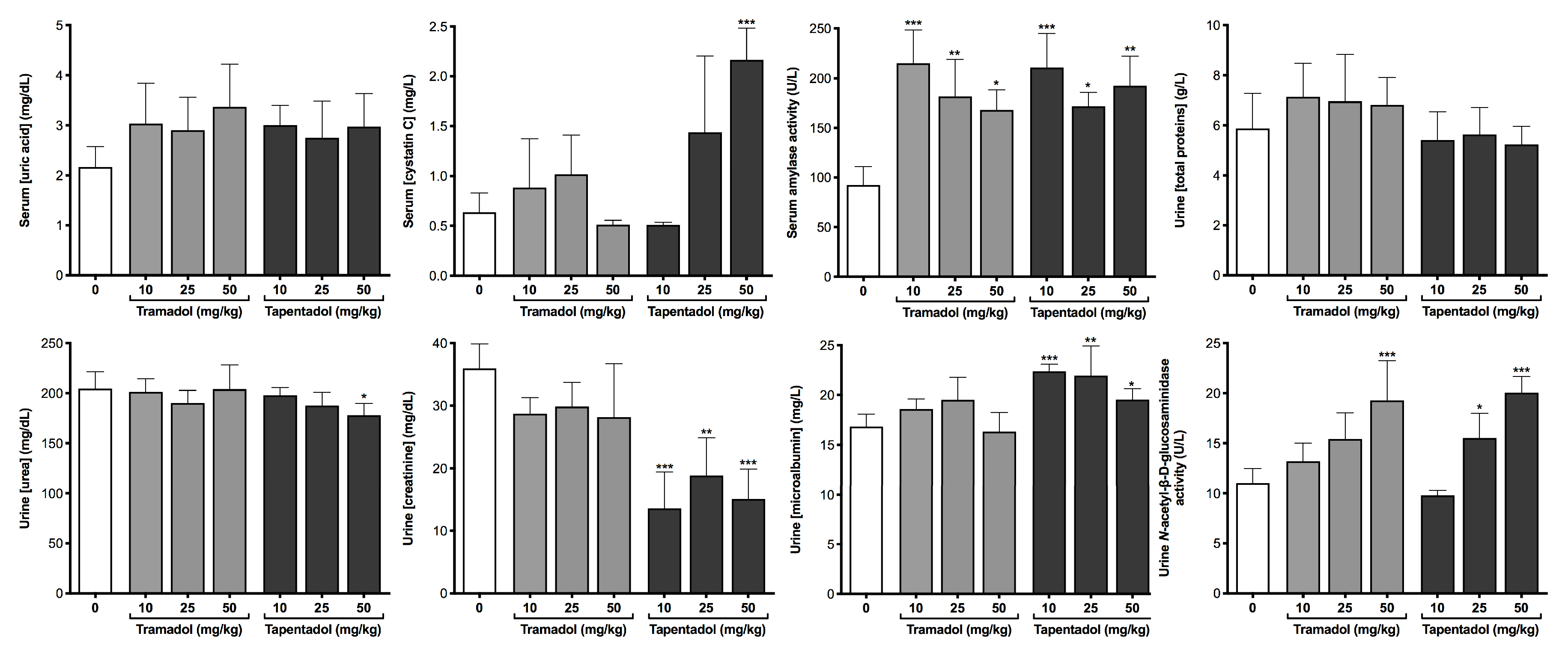
2.2. Repeated Exposure to Tramadol and Tapentadol Compromises Liver and Kidney Metabolic and Excretion Functions
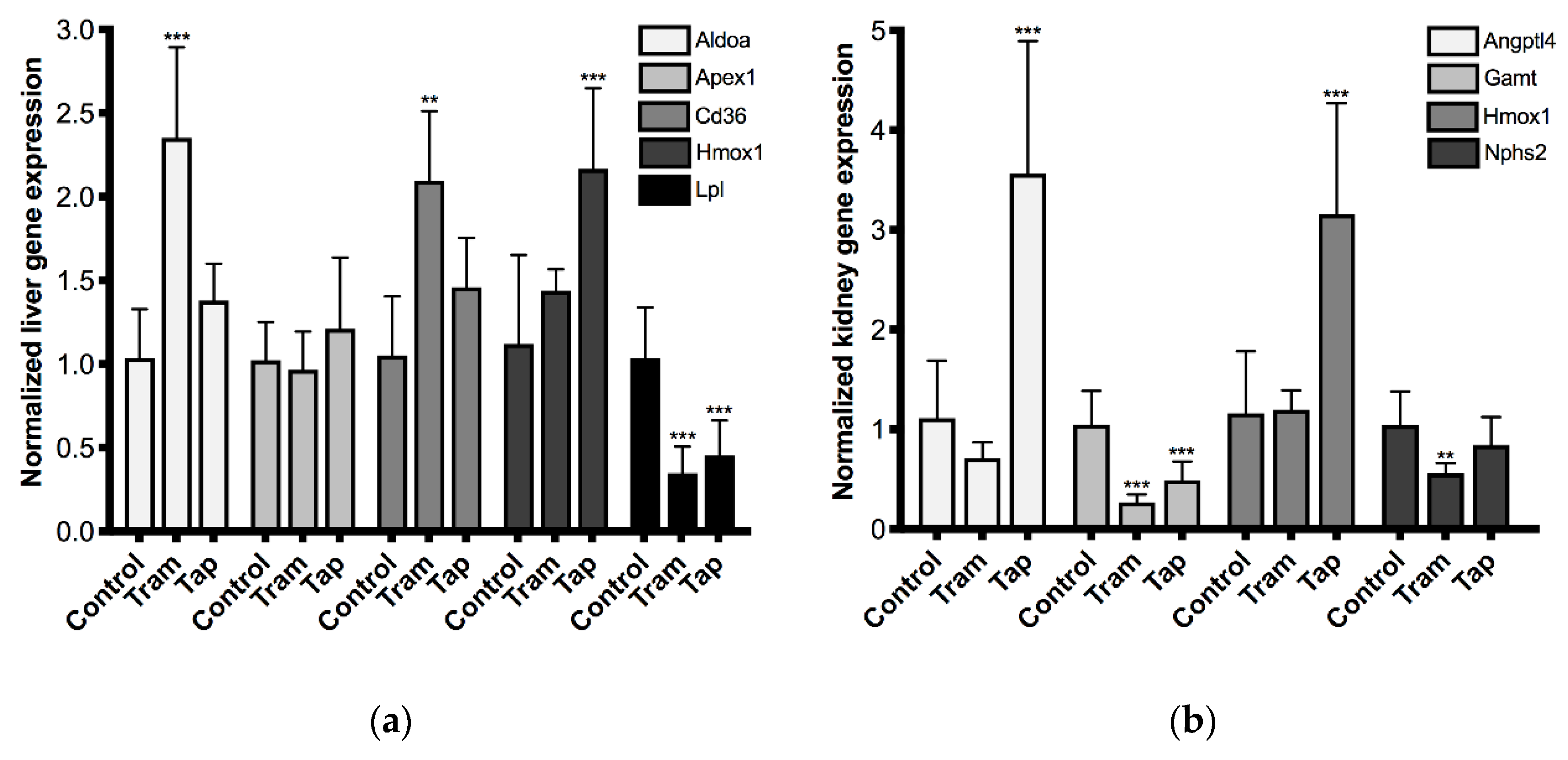
2.3. Repeated Exposure to Tramadol and Tapentadol Leads to Changes in the Gene Expression of Liver and Kidney Toxicity Biomarkers
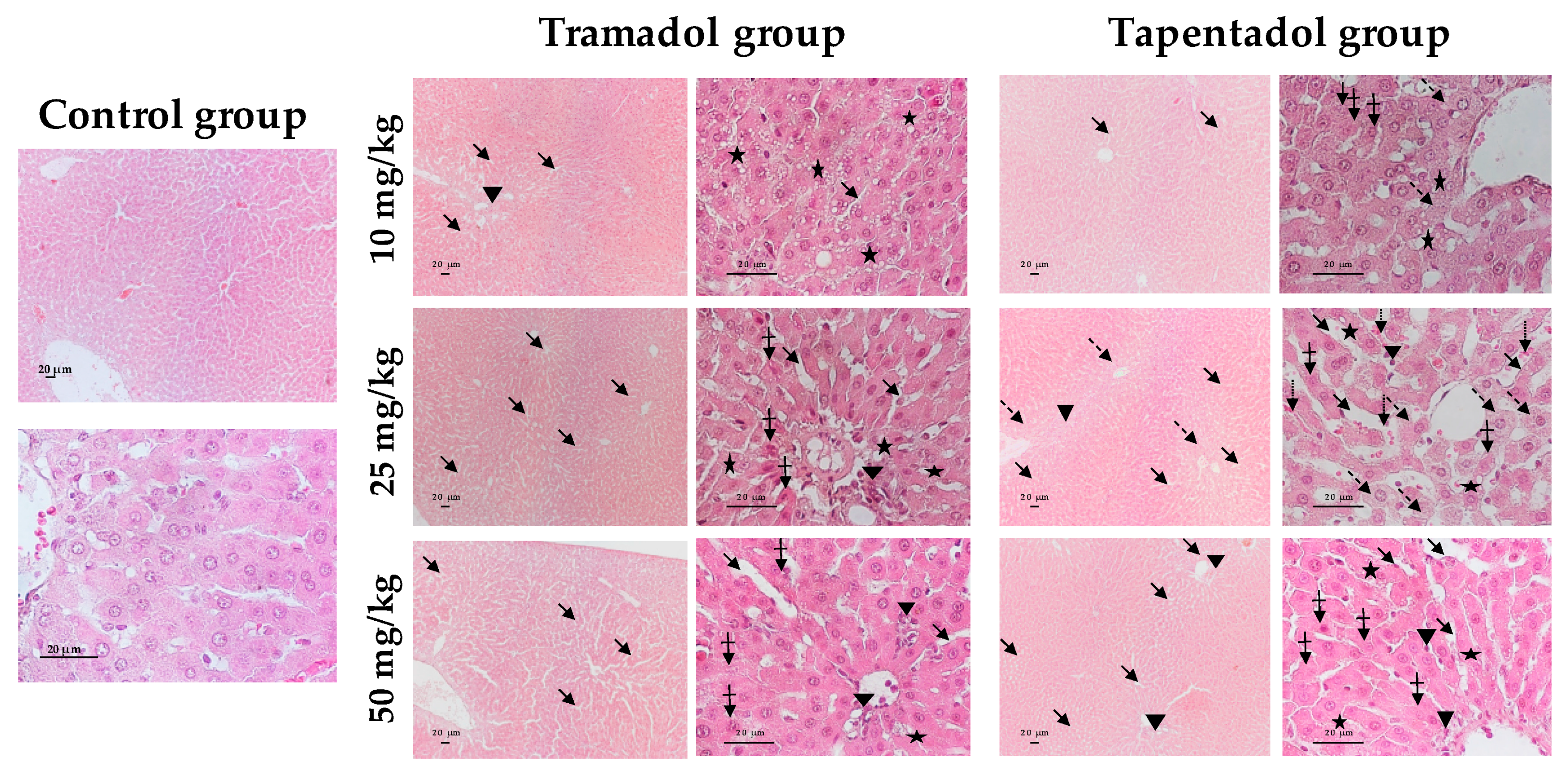
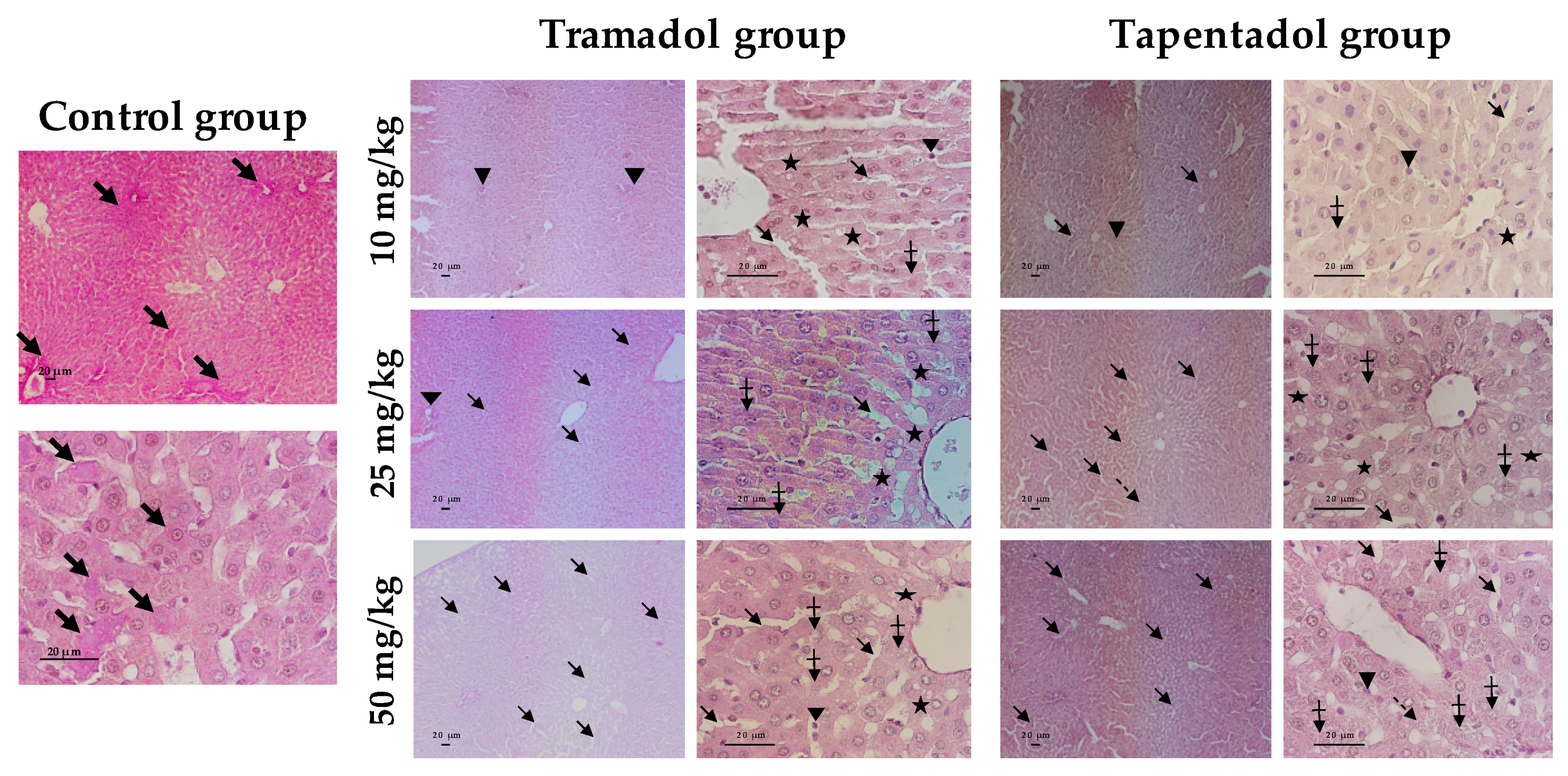
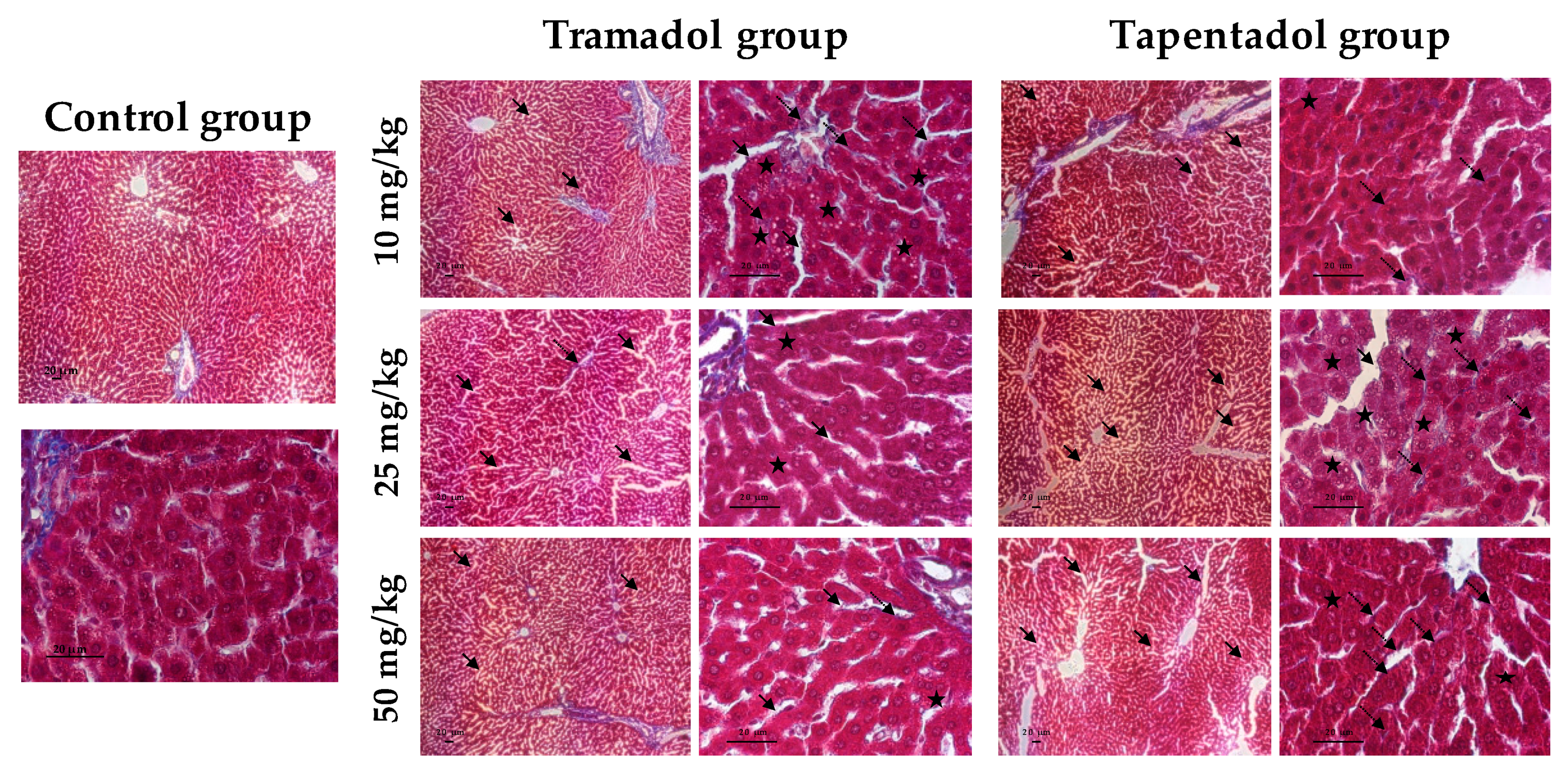
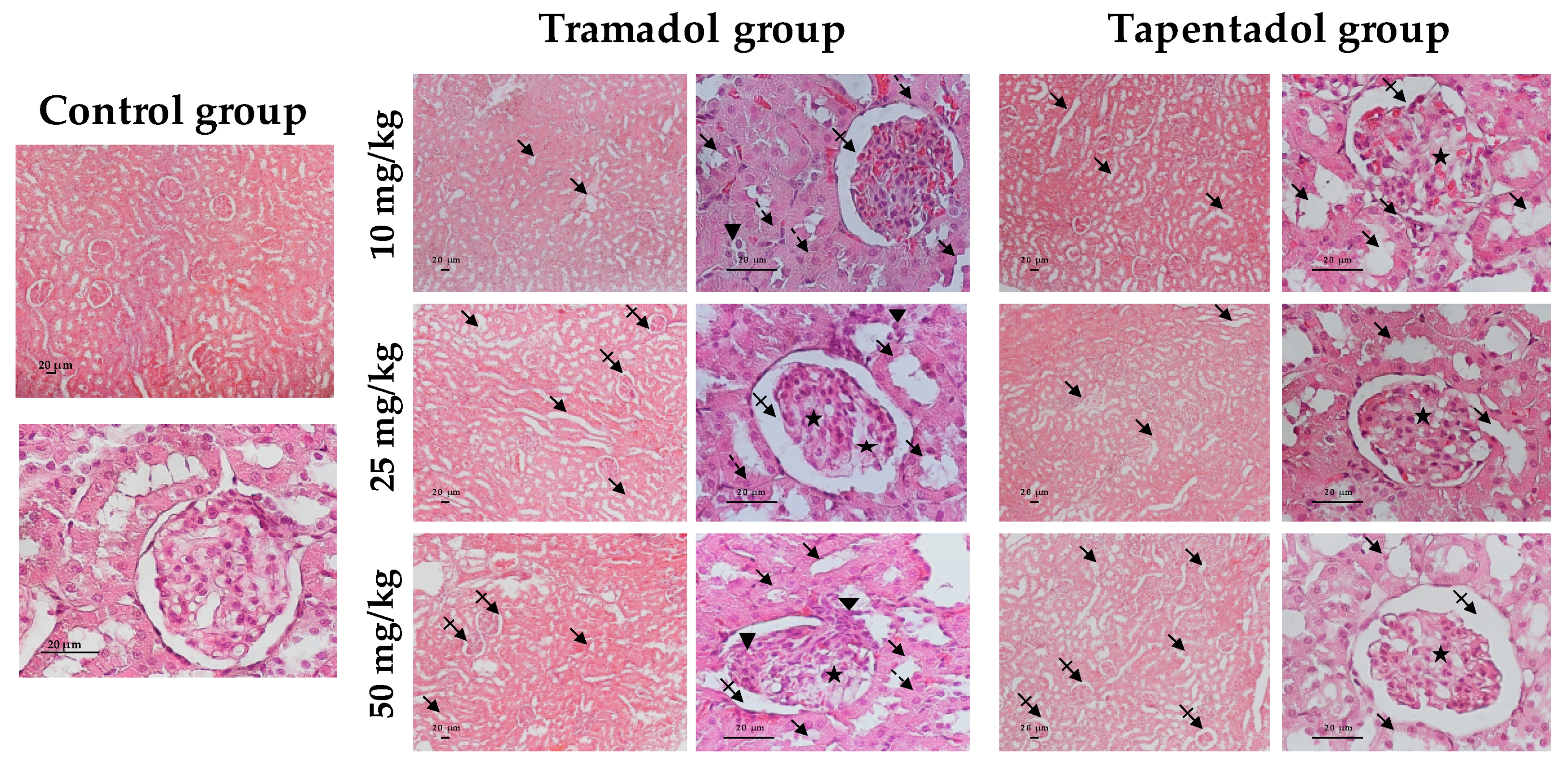
2.4. Repeated Exposure to Tramadol and Tapentadol Leads to Glycogen Depletion, Microsteatosis and Inflammation in Liver and Kidney, and to Fibrous Tissue Deposition between Hepatocytes
3. Discussion
3.1. Repeated Exposure to Tramadol and Tapentadol Induces Hepato-Renal Oxidative Stress, Affecting Liver and Kidney Cell Integrity and Function
3.2. Repeated Exposure to Tramadol and Tapentadol Causes Cumulative Hepatocellular and Hepatobiliary Damage
3.3. Repeated Exposure to Tramadol and Tapentadol Compromises Liver Synthesis
3.4. Repeated Exposure to Tramadol and Tapentadol Affects Lipid Profile, Correlating with Hepatobiliary Commitment and Lipid Deposition
3.5. Repeated Exposure to Tramadol and Tapentadol Affects Iron Metabolism, Correlating with Oxidative Stress, Cellular Damage, Inflammation and Steatosis
3.6. Repeated Exposure to Tramadol and Tapentadol Compromises Kidney Glomerular and Tubular Functions
3.7. Repeated Exposure to Tramadol and Tapentadol Alters Hepato-Renal Toxicity Biomarker Gene Expression, Correlating with Metabolic Changes, Cell Toxicity and Glomerular Dysfunction
3.8. Repeated Exposure to Tramadol and Tapentadol Causes Liver and Kidney Histopathological Changes, Correlating with Metabolic and Gene Expression Alterations
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Models and Animal Handling
4.3. Experimental Design and Drug Treatment
4.4. Collection and Processing of Biological Samples
4.4.1. Quantification of Oxidative Stress Parameters
4.4.2. Quantification of Biochemical Parameters in Serum and Urine Samples
4.4.3. Gene Expression Analysis through qRT-PCR
4.4.4. Liver and Kidney Histopathological Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faria, J.; Barbosa, M.J.; Moreira, R.; Queirós, O.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative pharmacology and toxicology of tramadol and tapentadol. Eur. J. Pain 2018, 22, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.J.; Faria, J.; Queirós, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative metabolism of tramadol and tapentadol: A toxicological perspective. Drug Metab. Rev. 2016, 48, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-C.; Chen, L.-C.; Kerry, M.; Knaggs, R. Prescription opioids: Regional variation and socioeconomic status—Evidence from primary care in England. Int. J. Drug Policy 2019, 64, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Curtis, H.J.; Croker, R.; Walker, A.J.; Richards, G.C.; Quinlan, J.; Goldacre, B. Opioid prescribing trends and geographical variation in England, 1998–2018: A retrospective database study. Lancet Psychiatry 2019, 6, 140–150. [Google Scholar] [CrossRef]
- Scholten, W.K.; Christensen, A.-E.; Olesen, A.E.; Drewes, A.M. Quantifying the Adequacy of Opioid Analgesic Consumption Globally: An Updated Method and Early Findings. Am. J. Public Health 2018, 109, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Bolshakova, M.; Bluthenthal, R.N.; Sussman, S. Opioid use and misuse: Health impact, prevalence, correlates and interventions. Psychol. Health 2019, 34, 1105–1139. [Google Scholar] [CrossRef] [PubMed]
- Chenaf, C.; Kaboré, J.-L.; Delorme, J.; Pereira, B.; Mulliez, A.; Zenut, M.; Delage, N.; Ardid, D.; Eschalier, A.; Authier, N. Prescription opioid analgesic use in France: Trends and impact on morbidity-mortality. Eur. J. Pain 2018, 23, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Tuminello, S.; Alpert, N.; Flores, R.; Taioli, E. Physician prescribing practices and opioid misuse in the USA. Lancet Psychiatry 2019, 6, e7. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, D.F.; Becker, W.C.; Fiellin, D.; Stannard, C. Prescription opioid misuse in the United States and the United Kingdom: Cautionary lessons. Int. J. Drug Policy 2014, 25, 1124–1130. [Google Scholar] [CrossRef]
- Murphy, D.L.; Lebin, J.A.; Severtson, S.G.; Olsen, H.A.; Dasgupta, N.; Dart, R.C. Comparative Rates of Mortality and Serious Adverse Effects Among Commonly Prescribed Opioid Analgesics. Drug Saf. 2018, 41, 787–795. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; LeQuang, J.A.; Taylor, R.; Ossipov, M.H.; Colucci, D.; Raffa, R.B. Designing safer analgesics: A focus on μ-opioid receptor pathways. Expert Opin. Drug Discov. 2018, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Ashaye, T.; Hounsome, N.; Carnes, D.; Taylor, S.J.C.; Homer, K.; Eldridge, S.; Spencer, A.; Rahman, A.; Foell, J.; Underwood, M.R. Opioid prescribing for chronic musculoskeletal pain in UK primary care: Results from a cohort analysis of the COPERS trial. BMJ Open 2018, 8, e019491. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Santucci, C.; Radrezza, S.; Erthal, J.; Berterame, S.; Corli, O. Trends in the consumption of opioids for the treatment of severe pain in Europe, 1990–2016. Eur. J. Pain 2018, 23, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedenmalm, K.; Slattery, J.; Skibicka-Stepien, I.; Kurz, X.; Morales, D. Prescribing patterns of tramadol in adults in IMS® primary care databases in France and Germany between 1 January 2006 and 30 June 2016. Eur. J. Clin. Pharmacol. 2019, 75, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Kalkman, G.A.; Kramers, C.; Van Dongen, R.T.; Brink, W.V.D.; Schellekens, A. Trends in use and misuse of opioids in the Netherlands: A retrospective, multi-source database study. Lancet Public Health 2019, 4, e498–e505. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Luo, C.; Dai, H.; Fang, W. Consumption trends and prescription patterns of opioids from 2011 to 2016: A survey in a Chinese city. BMJ Open 2019, 9, e021923. [Google Scholar] [CrossRef] [Green Version]
- Reset, A.; Skurtveit, S.; Furu, K.; Skovlund, E. Effect of the market withdrawal of dextropropoxyphene on use of other prescribed analgesics. Scand. J. Pain 2018, 18, 667–674. [Google Scholar] [CrossRef]
- Tanghe, M.; Van Den Noortgate, N.; Pivodic, L.; Deliens, L.; Onwuteaka-Philipsen, B.; Szczerbińska, K.; Finne-Soveri, H.; Collingridge-Moore, D.; Gambassi, G.; Van Den Block, L.; et al. Opioid, antipsychotic and hypnotic use in end of life in long-term care facilities in six European countries: Results of PACE. Eur. J. Public Health 2018, 29, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Bravo, L.; Mico, J.A.; Berrocoso, E. Discovery and development of tramadol for the treatment of pain. Expert Opin. Drug Discov. 2017, 12, 1281–1291. [Google Scholar] [CrossRef]
- Giorgi, M. Tramadol Vs Tapentadol: Anew Horizon in Pain Treatment? Am. J. Anim. Veter. Sci. 2012, 7, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; McTavish, D.; Sorkin, E.M. Tramadol. Drugs 1993, 46, 313–340. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, J.; Alegre, C.; Blake, D.; Alén, J.C.; Caporali, R.; Casser, H.; Correa-Illanes, G.; Fernandes, P.; Galilea, E.; Jány, R.; et al. Current Considerations for the Treatment of Severe Chronic Pain: The Potential for Tapentadol. Pain Pract. 2011, 12, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Power, I. An update on analgesics. Br. J. Anaesth. 2011, 107, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.R.; Nag, K.; Shetti, A.N.; Krishnaveni, N. Tapentadol hydrochloride: A novel analgesic. Saudi J. Anaesth. 2013, 7, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Tzschentke, T.M.; Christoph, T.; Kögel, B.Y. The Mu-Opioid Receptor Agonist/Noradrenaline Reuptake Inhibition (MOR–NRI) Concept in Analgesia: The Case of Tapentadol. CNS Drugs 2014, 28, 319–329. [Google Scholar] [CrossRef]
- Vadivelu, N.; Mitra, S.; Narayan, D. Recent Advances in Postoperative Pain Management. Yale J. Biol. Med. 2010, 83, 11–25. [Google Scholar]
- Ramaswamy, S.; Chang, S.; Mehta, V. Tapentadol—The evidence so far. Anaesthesia 2015, 70, 518–522. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Kataoka, T.; Tasaki, Y.; Kondo, Y.; Sato, N.; Naiki, T.; Sakamoto, N.; Akechi, T.; Kimura, K. Efficacy of tapentadol for first-line opioid-resistant neuropathic pain in Japan. Jpn. J. Clin. Oncol. 2018, 48, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Klose, P.; Welsch, P.; Petzke, F.; Häuser, W. Opioids for chronic non-cancer neuropathic pain. An updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks duration. Eur. J. Pain 2019, 24, 3–18. [Google Scholar] [CrossRef]
- Caraci, F.; Merlo, S.; Drago, F.; Caruso, G.; Parenti, C.; Sortino, M.A. Rescue of Noradrenergic System as a Novel Pharmacological Strategy in the Treatment of Chronic Pain: Focus on Microglia Activation. Front. Pharmacol. 2019, 10, 1024. [Google Scholar] [CrossRef] [Green Version]
- Kress, H.G.; Koch, E.D.; Kosturski, H.; Steup, A.; Karcher, K.; Dogan, C.; Etropolski, M.; Eerdekens, M. Direct conversion from tramadol to tapentadol prolonged release for moderate to severe, chronic malignant tumour-related pain. Eur. J. Pain 2016, 20, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, R.; Reuter, H. An overview of analgesics: Opioids, tramadol, and tapentadol (Part 2). S. Afr. Fam. Pract. 2019, 61, 16–23. [Google Scholar] [CrossRef]
- Vosburg, S.K.; Severtson, S.G.; Dart, R.C.; Cicero, T.J.; Kurtz, S.P.; Parrino, M.W.; Green, J.L. Assessment of Tapentadol API Abuse Liability With the Researched Abuse, Diversion and Addiction-Related Surveillance System. J. Pain 2018, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A.; Rose, M.A. The anaesthetist, opioid analgesic drugs, and serotonin toxicity: A mechanistic and clinical review. Br. J. Anaesth. 2019, 124, 44–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef]
- Raffa, R.B.; Buschmann, H.; Christoph, T.; Eichenbaum, G.; Englberger, W.; Flores, C.M.; Hertrampf, T.; Kögel, B.; Schiene, K.; Straßburger, W.; et al. Mechanistic and functional differentiation of tapentadol and tramadol. Expert Opin. Pharmacother. 2012, 13, 1437–1449. [Google Scholar] [CrossRef]
- Leppert, W. CYP2D6 in the Metabolism of Opioids for Mild to Moderate Pain. Pharmacology 2011, 87, 274–285. [Google Scholar] [CrossRef]
- Wu, F.; Slawson, M.H.; Johnson-Davis, K.L. Metabolic Patterns of Fentanyl, Meperidine, Methylphenidate, Tapentadol and Tramadol Observed in Urine, Serum or Plasma. J. Anal. Toxicol. 2017, 41, 289–299. [Google Scholar] [CrossRef]
- DePriest, A.Z.; Puet, B.L.; Holt, A.C.; Roberts, A.; Cone, E.J. Metabolism and Disposition of Prescription Opioids: A Review. Forensic Sci. Rev. 2015, 27, 115–145. [Google Scholar]
- Chang, E.J.; Choi, E.J.; Kim, K.H. Tapentadol: Can It Kill Two Birds with One Stone without Breaking Windows? Korean J. Pain 2016, 29, 153–157. [Google Scholar] [CrossRef]
- Langford, R.M.; Knaggs, R.; Farquhar-Smith, P.; Dickenson, A.H. Is tapentadol different from classical opioids? A review of the evidence. Br. J. Pain 2016, 10, 217–221. [Google Scholar] [CrossRef]
- Channell, J.S.; Schug, S. Toxicity of tapentadol: A systematic review. Pain Manag. 2018, 8, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Raffa, R.B.; Elling, C.; Tzschentke, T.M. Does ‘Strong Analgesic’ Equal ‘Strong Opioid’? Tapentadol and the Concept of ‘µ-Load’. Adv. Ther. 2018, 35, 1471–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartrick, C.T.; Rozek, R.J. Tapentadol in Pain Management. CNS Drugs 2011, 25, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Kneip, C.; Terlinden, R.; Beier, H.; Chen, G. Investigations into the drug-drug interaction potential of tapentadol in human liver microsomes and fresh human hepatocytes. Drug Metab. Lett. 2008, 2, 67–75. [Google Scholar] [CrossRef]
- Terlinden, R.; Ossig, J.; Fliegert, F.; Lange, C.; Göhler, K. Absorption, metabolism, and excretion of 14C-labeled tapentadol HCl in healthy male subjects. Eur. J. Drug Metab. Pharmacokinet. 2007, 32, 163–169. [Google Scholar] [CrossRef]
- Borys, D.; Stanton, M.; Gummin, D.; Drott, T. Tapentadol Toxicity in Children. Pediatrics 2015, 135, 392–396. [Google Scholar] [CrossRef] [Green Version]
- Karila, L.; Marillier, M.; Chaumette, B.; Billieux, J.; Franchitto, N.; Benyamina, A.; Nicolas, F.; Amine, B. New synthetic opioids: Part of a new addiction landscape. Neurosci. Biobehav. Rev. 2019, 106, 133–140. [Google Scholar] [CrossRef]
- Suga, Y.; Uchida, M.; Suzuki, S.; Sugawara, H.; Torigoe, K.; Futamura, A.; Uesawa, Y.; Nakagawa, T.; Takase, H. Current Status of Adverse Events Related with Opioid Analgesics in Japan: Assessment Based on Japanese Adverse Drug Event Report Database. Biol. Pharm. Bull. 2019, 42, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Oliveira, A.; Costa, I.S.B.; Gouveia, C.A.; Carvalho, F.; Moreira, R.F.; Dinis-Oliveira, R.J. Simultaneous quantification of tramadol and O-desmethyltramadol in hair samples by gas chromatography-electron impact/mass spectrometry. Biomed. Chromatogr. 2013, 27, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Tchoe, H.J.; Li, J.; Shin, J.-Y. All-Cause Mortality Associated with Tramadol Use: A Case-Crossover Study. Drug Saf. 2019, 42, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, F.L.; Mallett, P.; Aldridge, L.; Verilhac, K.; McIntyre, I.M. A tapentadol related fatality: Case report with postmortem concentrations. Forensic Sci. Int. 2016, 266, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.S.B.; Oliveira, A.; De Pinho, P.G.; Teixeira, H.M.; Moreira, R.F.; Carvalho, F.; Dinis-Oliveira, R.J. Postmortem Redistribution of Tramadol and O-Desmethyltramadol. J. Anal. Toxicol. 2013, 37, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.M.; Ali, Z.; Levine, B.; Middleberg, R.A.; Fowler, D.R. Case Report of a Fatal Intoxication by Nucynta. Am. J. Forensic Med. Pathol. 2014, 35, 234–236. [Google Scholar] [CrossRef]
- Hawton, K.; Ferrey, A.; Casey, D.; Wells, C.; Fuller, A.; Bankhead, C.; Clements, C.; Ness, J.; Gunnell, D.; Kapur, N.; et al. Relative toxicity of analgesics commonly used for intentional self-poisoning: A study of case fatality based on fatal and non-fatal overdoses. J. Affect. Disord. 2019, 246, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Kemp, W.L.; Schlueter, S.; Smalley, E. Death Due to Apparent Intravenous Injection of Tapentadol. J. Forensic Sci. 2012, 58, 288–291. [Google Scholar] [CrossRef]
- Khaja, M.; Lominadze, G.; Millerman, K. Cardiac Arrest Following Drug Abuse with Intravenous Tapentadol: Case Report and Literature Review. Am. J. Case Rep. 2017, 18, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Larson, S.J.; Pestaner, J.; Prashar, S.K.; Bayard, C.; Zarwell, L.W.; Pierre-Louis, M. Postmortem Distribution of Tapentadol and N-Desmethyltapentadol. J. Anal. Toxicol. 2012, 36, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Loughrey, M.; Loughrey, C.; Johnston, S.; O’Rourke, D. Fatal hepatic failure following accidental tramadol overdose. Forensic Sci. Int. 2003, 134, 232–233. [Google Scholar] [CrossRef]
- Partridge, E.; Teoh, E.; Nash, C.; Scott, T.; Charlwood, C.; Kostakis, C. The Increasing Use and Abuse of Tapentadol and Its Incorporation Into a Validated Quantitative Method. J. Anal. Toxicol. 2018, 42, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Pilgrim, J.L.; Gerostamoulos, D.; Drummer, O.H. Deaths involving contraindicated and inappropriate combinations of serotonergic drugs. Int. J. Leg. Med. 2010, 125, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, J.L.; Gerostamoulos, D.; Drummer, O.H. Deaths involving serotonergic drugs. Forensic Sci. Int. 2010, 198, 110–117. [Google Scholar] [CrossRef]
- Tjäderborn, M.; Jönsson, A.K.; Hägg, S.; Ahlner, J. Fatal unintentional intoxications with tramadol during 1995–2005. Forensic Sci. Int. 2007, 173, 107–111. [Google Scholar] [CrossRef]
- Barbera, N.G.E.; Fisichella, M.; Bosco, A.; Indorato, F.; Spadaro, G.; Romano, G. A suicidal poisoning due to tramadol. A metabolic approach to death investigation. J. Forensic Leg. Med. 2013, 20, 555–558. [Google Scholar] [CrossRef] [PubMed]
- De Backer, B.; Renardy, F.; Denooz, R.; Charlier, C. Quantification in postmortem blood and identification in urine of tramadol and its two main metabolites in two cases of lethal tramadol intoxication. J. Anal. Toxicol. 2010, 34, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musshoff, F.; Madea, B. Fatality due to ingestion of tramadol alone. Forensic Sci. Int. 2001, 116, 197–199. [Google Scholar] [CrossRef]
- Lusthof, K.J.; Zweipfenning, P.G. Suicide by Tramadol Overdose. J. Anal. Toxicol. 1998, 22, 260. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.A.; Cina, S.J.; Jones, R.; Selby, D.M.; Levine, B.; Smith, M.L. Tissue distribution of tramadol and metabolites in an overdose fatality. Am. J. Forensic Med. Pathol. 1999, 20, 98–100. [Google Scholar] [CrossRef]
- Rickli, A.; Liakoni, E.; Hoener, M.C.; Liechti, M.E. Opioid-induced inhibition of the human 5-HT and noradrenaline transporters in vitro: Link to clinical reports of serotonin syndrome. Br. J. Pharmacol. 2018, 175, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, P.; Pakhre, A.; Kattula, D.; Sarkar, S. Tapentadol Dependence: A Case Series. Prim. Care Companion CNS Disord. 2019, 21. [Google Scholar] [CrossRef]
- Atici, Ş.; Cinel, I.; Cinel, L.; Doruk, N.; Eskandari, H.G.; Oral, U. Liver and kidney toxicity in chronic use of opioids: An experimental long term treatment model. J. Biosci. 2005, 30, 245–252. [Google Scholar] [CrossRef] [PubMed]
- El Fatoh, M.F.; Farag, M.R.; Sayed, S.A.E.; Kamel, M.A.; Abdel-Hamid, N.E.; Hussein, M.A.; Salem, G.A. Some biochemical, neurochemical, pharmacotoxicological and histopathological alterations induced by long-term administration of tramadol in male rats. Int. J. Pharm. Sci. 2014, 4, 565–571. [Google Scholar]
- Albarakai, A.Y.; Alsbery, H.M.A.E. Evaluation of the hepatoprotective efficacy of Moringa oleifera on Tramal-induced liver toxicity in animal models. Res. J. Pharm. Biol. Chem. Sci. 2016, 7, 1494–1501. [Google Scholar]
- Ali, O.K.; Ahmed, A.-J.S.; Mawlood, A.-G. Effects of tramadol on histopathological and biochemical parameters in male rabbits. Am. J. Biol. Life Sci. 2015, 3, 85–90. [Google Scholar]
- El-Gaafarawi, I.I. Biochemical toxicity induced by tramadol administration in male rats. Egypt J. Hosp. Med. 2006, 23, 353–362. [Google Scholar]
- Elkhateeb, A.; El Khishin, I.; Megahed, O.H.; Mazen, F. Effect of Nigella sativa Linn oil on tramadol-induced hepato- and nephrotoxicity in adult male albino rats. Toxicol. Rep. 2015, 2, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Ezzeldin, E.; Souror, W.A.H.; El-Nahhas, T.; Soudi, A.N.M.M.; Shahat, A.A. Biochemical and Neurotransmitters Changes Associated with Tramadol in Streptozotocin-Induced Diabetes in Rats. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Hafez, E.; Issa, S.; Rahman, S.A. Parenchymatous toxicity of tramadol: Histopathological and biochemical study. J. Alcohol Drug Depend. 2015, 3, 5. [Google Scholar]
- Saleem, R.; Iqbal, R.; Abbas, M.N.; Zahra, A.; Iqbal, J.; Ansari, M.S. Effects of tramadol on histopathological and biochemical parameters in mice (Mus musculus) model. Glob. J. Pharmacol. 2014, 8, 14–19. [Google Scholar]
- Samarghandian, S.; Afshari, R.; Farkhondeh, T. Effect of long-term treatment of morphine on enzymes, oxidative stress indices and antioxidant status in male rat liver. Int. J. Clin. Exp. Med. 2014, 7, 1449–1453. [Google Scholar]
- Youssef, H.S.; Azza, Z.H.M. Histopathological and biochemical effects of acute & chronic tramadol drug toxicity on liver, kidney and testicular function in adult male albino rats. Forensic Res. Criminol. Int. J. 2016, 2, 138–144. [Google Scholar]
- Zhang, Y.-T.; Zheng, Q.; Pan, J.; Zheng, R.-L. Oxidative Damage of Biomolecules in Mouse Liver Induced by Morphine and Protected by Antioxidants. Pharmacol. Toxicol. 2004, 95, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.A.; Ismail, H.K.; Aal, S.A. Effect of tramadol drug on some biochemical and immunological parameters in albino male rats; Evaluation of possible reversal following its withdrawal. Benha Veter. Med. J. 2017, 33, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.; Ibrahim, H.M.; Mohamed, A.A.; Tammam, H.G.; Mohammed, A.A. Vitamin E supplementation ameliorates the hepatotoxicity induced by Tramadol: Toxicological, histological and immunohistochemical study. Toxicol. Mech. Methods 2019, 30, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Albarakai, A. Histopathological, Biochemical and Haematological Changes of Nalbuphine-Hcl Administration on Liver of Albino Rat. Int. J. Adv. Res. 2017, 5, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Elyajzi, N.R.; Abdel-Aziz, I.; Aldalou, A.; Shahwan, O. The Effects of Tramadol Hydrochloride Administration on the Hematological and Biochemical Profiles of domestic male Rabbits. IUG J. Nat. Eng. Stud. 2013, 21, 51–65. [Google Scholar]
- Salahshoor, M.R.; Khashiadeh, M.; Roshankhah, S.; Kakabaraei, S.; Jalili, C. Protective effect of crocin on liver toxicity induced by morphine. Res. Pharm. Sci. 2016, 11, 120–129. [Google Scholar]
- Awadalla, E.A.; Salah-Eldin, A.E. Histopathological and molecular studies on tramadol mediated hepato-renal toxicity in rats. IOSR J. Pharm. Biol. Sci. 2015, 10, 90–102. [Google Scholar]
- Awadalla, E.A.; Salah-Eldin, A.-E. Molecular and histological changes in cerebral cortex and lung tissues under the effect of tramadol treatment. Biomed. Pharmacother. 2016, 82, 269–280. [Google Scholar] [CrossRef]
- Othman, G.Q. Evaluation of Oxidative Markers, Apoptosis and Reproductive Efficiency in Heroin Addicted Rats. IOSR J. Pharm. (IOSRPHR) 2013, 3, 1–7. [Google Scholar] [CrossRef]
- Payabvash, S.; Beheshtian, A.; Salmasi, A.H.; Kiumehr, S.; Ghahremani, M.H.; Tavangar, S.M.; Sabzevari, O.; Dehpour, A.R. Chronic morphine treatment induces oxidant and apoptotic damage in the mice liver. Life Sci. 2006, 79, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Kaoud, H.A.; Hellal, M.H.; Malhat, F.M.; Saeid, S.; Elmawella, I.A.; Khali, A.H. Effects of acute sub-lethal dose of tramadol on α2-adrenergic receptors and liver histopathology in rat. Glob. J. Curr. Res. 2013, 1, 70–76. [Google Scholar]
- Rabei, H.M. The immunological and histopathological changes of tramadol, tramadol/acetaminophen and acetaminophen in male albino rats—Comparative study. Egypt J. Hosp. Med. 2011, 45, 477–503. [Google Scholar]
- Samaka, R.M.; Girgis, N.F.; Shams, T.M. Acute toxicity and dependence of tramadol in albino rats: Relationship of Nestin and Notch 1 as stem cell markers. J. Am. Sci. 2012, 8, 313–327. [Google Scholar]
- Al-Mashhadane, F.A.; Ismail, H.K.; Al-Saidya, A.M. Histopathological effects of chronic use of tramadol on liver and kidney in sheep model. J. Pharm. Sci. Res. 2019, 11, 2208–2212. [Google Scholar]
- Borzelleca, J.F.; Egle, J.L.; Harris, L.S.; Johnson, D.N.; Terrill, J.B.; Belleville, J.A.N. Toxicological evaluation of μ-agonists part I: Assessment of toxicity following 30 days of repeated oral dosing of male and female rats with levo-alpha-acetylmethadol HCl (LAAM). J. Appl. Toxicol. 1994, 14, 435–446. [Google Scholar] [CrossRef]
- Faria, J.; Barbosa, M.J.; Queirós, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative study of the neurotoxicological effects of tramadol and tapentadol in SH-SY5Y cells. Toxicology 2016, 359, 1–10. [Google Scholar] [CrossRef]
- Barbosa, M.J.; Faria, J.; Leal, S.; Afonso, L.P.; Lobo, J.; Queirós, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Acute administration of tramadol and tapentadol at effective analgesic and maximum tolerated doses causes hepato- and nephrotoxic effects in Wistar rats. Toxicology 2017, 389, 118–129. [Google Scholar] [CrossRef]
- Faria, J.; Barbosa, M.J.; Leal, S.; Afonso, L.P.; Lobo, J.; Moreira, R.; Queirós, O.; Carvalho, F.; Dinis-Oliveira, R.J. Effective analgesic doses of tramadol or tapentadol induce brain, lung and heart toxicity in Wistar rats. Toxicology 2017, 385, 38–47. [Google Scholar] [CrossRef]
- Thoolen, B.; Maronpot, R.R.; Harada, T.; Nyska, A.; Rousseaux, C.; Nolte, T.; Malarkey, D.E.; Kaufmann, W.; Küttler, K.; Deschl, U.; et al. Proliferative and Nonproliferative Lesions of the Rat and Mouse Hepatobiliary System. Toxicol. Pathol. 2010, 38, 5S–81S. [Google Scholar] [CrossRef]
- Lukas, G.; Brindle, S.D.; Greengard, P. The route of absorption of intraperitoneally administered compounds. J. Pharmacol. Exp. Ther. 1971, 178, 562–564. [Google Scholar] [PubMed]
- Elmanama, A.A.; Essawaf, H.N.; Abu Tayyem, N.E.S. Tramadol-Induced Liver and Kidney Toxicity among Abusers in Gaza Strip, Palestine. Jordan J. Biol. Sci. 2015, 8, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; James, L.; Shi, Q.; Salminen, W.F. Hepatic toxicity biomarkers. In Biomarkers in Toxicology; Elsevier BV: Amsterdam, The Netherlands, 2014; pp. 241–259. [Google Scholar]
- Tang, N.; Zhang, Y.; Liu, Z.; Fu, T.; Liang, Q.; Ai, X. Correlation analysis between four serum biomarkers of liver fibrosis and liver function in infants with cholestasis. Biomed. Rep. 2016, 5, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallagangula, K.S.; Nagaraj, S.K.; Venkataswamy, L.; Chandrappa, M. Liver fibrosis: A compilation on the biomarkers status and their significance during disease progression. Futur. Sci. OA 2018, 4, FSO250. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.M.; Mahmoud, A.M. Chronic exposure to the opioid tramadol induces oxidative damage, inflammation and apoptosis, and alters cerebral monoamine neurotransmitters in rats. Biomed. Pharmacother. 2019, 110, 239–247. [Google Scholar] [CrossRef]
- Santarpia, L.; Grandone, I.; Contaldo, F.; Pasanisi, F. Butyrylcholinesterase as a prognostic marker: A review of the literature. J. Cachex Sarcopenia Muscle 2012, 4, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Asadikaram, G.; Reisi, M.; Kaseb, A.A.; Khaksari, M.; Mohammadi, A.; Mahmoodi, M. Effects of opium addiction on some serum factors in addicts with non-insulin-dependent diabetes mellitus. Addict. Biol. 2004, 9, 53–58. [Google Scholar] [CrossRef]
- Hebert, L.A.; Cosio, F.G.; Neff, J.C. Diagnostic significance of hypocomplementemia. Kidney Int. 1991, 39, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Weisman, S.; Goldsmith, B.; Winzler, R.; Lepper, M.H. Turnover of plasma orosomucoid in man. J. Lab. Clin. Med. 1961, 57, 7–15. [Google Scholar]
- Kuribayashi, T.; Seita, T.; Momotani, E.; Yamazaki, S.; Hagimori, K.; Yamamoto, S. Elimination Half-Lives of Acute Phase Proteins in Rats and Beagle Dogs During Acute Inflammation. Inflammation 2015, 38, 1401–1405. [Google Scholar] [CrossRef]
- Carpenter, C.B.; Ruddy, S.; Shehadeh, I.H.; Müller-Eberhard, H.J.; Merrill, J.P.; Austen, K.F. Complement metabolism in man: Hypercatabolism of the fourth (C4) and third (C3) components in patients with renal allograft rejection and hereditary angioedema (HAE). J. Clin. Investig. 1969, 48, 1495–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffay, H. The metabolism of serum proteins. 3. Kinetics of serum protein metabolism during growth. J. Biol. Chem. 1960, 235, 2352–2356. [Google Scholar] [PubMed]
- Zbarsky, S.H.; Wright, W.D. The Metabolism of C14-Urea in the Rat. Can. J. Med Sci. 1953, 31, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Najafipour, H.; Beik, A. The Impact of Opium Consumption on Blood Glucose, Serum Lipids and Blood Pressure, and Related Mechanisms. Front. Physiol. 2016, 7, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoudkabir, F.; Sarrafzadegan, N.; Eisenberg, M.J. Effects of opium consumption on cardiometabolic diseases. Nat. Rev. Cardiol. 2013, 10, 733–740. [Google Scholar] [CrossRef]
- Abs, R.; Verhelst, J.; Maeyaert, J.; Van Buyten, J.-P.; Opsomer, F.; Adriaensen, H.; Verlooy, J.; Van Havenbergh, T.; Smet, M.; Van Acker, K. Endocrine Consequences of Long-Term Intrathecal Administration of Opioids. J. Clin. Endocrinol. Metab. 2000, 85, 2215–2222. [Google Scholar] [CrossRef]
- Mohammadi, A.; Oshaghi, E.A.; Sorkhani, A.N.; Oubari, F.; Kia, R.H.; Rezaei, A.; Abbas, M.; Abbasi, O.E.; Noori, S.A.; Farhad, O.; et al. Effect of Opium on Lipid Profile and Expression of Liver X Receptor Alpha (LXRα) in Normolipidemic Mouse. Food Nutr. Sci. 2012, 3, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Najafipour, H.; Joukar, S.; Malekpour-Afshar, R.; Mirzaeipour, F.; Nasri, H. Passive opium smoking does not have beneficial effect on plasma lipids and cardiovascular indices in hypercholesterolemic rabbits with ischemic and non-ischemic hearts. J. Ethnopharmacol. 2010, 127, 257–263. [Google Scholar] [CrossRef]
- Sadeghian, S.; Boroumand, M.A.; Anvari, M.S.; Rabbani, S.; Sheikhfathollahi, M.; Abbasi, A. Effect of opium on glucose metabolism and lipid profiles in rats with streptozotocin-induced diabetes. Endokrynol. Polska 2009, 60, 258–262. [Google Scholar]
- Bryant, H.U.; Story, J.A.; Yim, G.K. Morphine-induced alterations in plasma and tissue cholesterol levels. Life Sci. 1987, 41, 545–554. [Google Scholar] [CrossRef]
- Mami, S.; Eghbali, M.; Cheraghi, J.; Mami, F.; Pourmahdi, B.M.; Salati, A.P. Effect of opium addiction on some serum parameters in rabbit. Glob. Vet. 2011, 7, 310–314. [Google Scholar]
- Mohammadi, A.; Darabi, M.; Nasry, M.; Saabet-Jahromi, M.-J.; Malek-Pour-Afshar, R.; Sheibani, H. Effect of opium addiction on lipid profile and atherosclerosis formation in hypercholesterolemic rabbits. Exp. Toxicol. Pathol. 2009, 61, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Mirzaei, F.; Jamshidi, M.; Yari, R.; Pak, S.; Sorkhani, A.N.; Norouzian, P.; Abdolkarimi, V.; Abbasi-Oshaghi, E. The In vivo Biochemical and Oxidative Changes by Ethanol and Opium Consumption in Syrian Hamsters. Int. J. Biol. 2013, 5, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Al Sagair, O.A. Effect of morphine sulphate on total lipids and triglycerides contents in serum and brain regions of rat. Med. Islam. World Sci. 2005, 15, 117–125. [Google Scholar]
- Bryant, H.U.; Kuta, C.C.; Story, J.A.; Yim, G.K. Stress- and morphine-induced elevations of plasma and tissue cholesterol in mice: Reversal by naltrexone. Biochem. Pharmacol. 1988, 37, 3777–3780. [Google Scholar] [CrossRef]
- Rahimi, N.; Gozashti, M.H.; Najafipour, H.; Shokoohi, M.; Marefati, H. Potential Effect of Opium Consumption on Controlling Diabetes and Some Cardiovascular Risk Factors in Diabetic Patients. Addict. Health 2014, 6, 1–6. [Google Scholar]
- Hillard, C.J. Lipids and drugs of abuse. Life Sci. 2005, 77, 1531–1542. [Google Scholar] [CrossRef]
- Ersche, K.D.; Acosta-Cabronero, J.; Jones, P.S.; Ziauddeen, H.; Van Swelm, R.P.L.; Laarakkers, C.M.M.; Raha-Chowdhury, R.; Williams, G.B. Disrupted iron regulation in the brain and periphery in cocaine addiction. Transl. Psychiatry 2017, 7, e1040. [Google Scholar] [CrossRef] [Green Version]
- Burhans, M.S.; Dailey, C.; Beard, Z.; Wiesinger, J.; Murray-Kolb, L.; Jones, B.C.; Beard, J.L. Iron deficiency: Differential effects on monoamine transporters. Nutr. Neurosci. 2005, 8, 31–38. [Google Scholar] [CrossRef]
- Immenschuh, S.; Baumgart-Vogt, E.; Mueller, S. Heme oxygenase-1 and iron in liver inflammation: A complex alliance. Curr. Drug Targets 2010, 11, 1541–1550. [Google Scholar] [CrossRef]
- Sass, G.; Barikbin, R.; Tiegs, G. The Multiple Functions of Heme Oxygenase-1 in the Liver. Z. Gastroenterol. 2012, 50, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Gautam, V.; Naseem, S. Acute-phase proteins: As diagnostic tool. J. Pharm. Bioallied Sci. 2011, 3, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Jurado, R.L. Iron, infections, and anemia of inflammation. Clin. Infect. Dis. 1997, 25, 888–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, N.; Takasawa, K. Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease. Nutrients 2018, 10, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Bhatt, L.; Horgan, C.P.; McCaffrey, M.W. Knockdown of β2-microglobulin perturbs the subcellular distribution of HFE and hepcidin. Biochem. Biophys. Res. Commun. 2009, 378, 727–731. [Google Scholar] [CrossRef]
- Li, L.; Dong, M.; Wang, X.-G. The Implication and Significance of Beta 2 Microglobulin. Chin. Med. J. 2016, 129, 448–455. [Google Scholar] [CrossRef]
- Zeng, X.; Hossain, D.; Bostwick, D.G.; Herrera, G.A.; Zhang, P.L. Urinary β2-Microglobulin Is a Good Indicator of Proximal Tubule Injury: A Correlative Study with Renal Biopsies. J. Biomark. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Drueke, T.B.; Massy, Z.A. Beta2-Microglobulin. Semin. Dial. 2009, 22, 378–380. [Google Scholar] [CrossRef]
- Liu, X.; Guan, Y.; Xu, S.; Li, Q.; Sun, Y.; Han, R.; Jiang, C. Early Predictors of Acute Kidney Injury: A Narrative Review. Kidney Blood Press. Res. 2016, 41, 680–700. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, C.P.; Chen, S.S.; Ng, Y.-H.; Roumelioti, M.-E.; Shaffi, K.; Singh, P.P.; Tzamaloukas, A.H. Rediscovering Beta-2 Microglobulin As a Biomarker across the Spectrum of Kidney Diseases. Front. Med. 2017, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Barton, K.T.; Kakajiwala, A.; Dietzen, D.J.; Goss, C.W.; Gu, H.; Dharnidharka, V.R. Using the newer kidney Disease: Improving global outcomes criteria, beta-2-microglobulin levels associate with severity of acute kidney injury. Clin. Kidney J. 2018, 11, 797–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethea, M.; Forman, D.T. Beta 2-microglobulin: Its significance and clinical usefulness. Ann. Clin. Lab. Sci. 1990, 20, 163–168. [Google Scholar] [PubMed]
- Muckenthaler, M.U.; Rodrigues, P.; Macedo, M.G.; Miñana, B.; Brennan, K.; Cardoso, E.M.; Hentze, M.W.; De Sousa, M. Molecular analysis of iron overload in β2-microglobulin-deficient mice. Blood Cells Mol. Dis. 2004, 33, 125–131. [Google Scholar] [CrossRef]
- Rodrigues, P.; Lopes, C.; Mascarenhas, C.; Arosio, P.; Porto, G.; De Sousa, M. Comparative study between Hfe−/− and β2m−/− mice: Progression with age of iron status and liver pathology. Int. J. Exp. Pathol. 2006, 87, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Viveiros, A.; Finkenstedt, A.; Schaefer, B.; Mandorfer, M.; Scheiner, B.; Lehner, K.; Tobiasch, M.; Reiberger, T.; Tilg, H.; Edlinger, M.; et al. Transferrin as a predictor of survival in cirrhosis. Liver Transpl. 2018, 24, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef]
- Abraham, N.; Cao, J.; Sacerdoti, D.; Li, X.; Drummond, G. Heme oxygenase: The key to renal function regulation. Am. J. Physiol. Renal Physiol. 2009, 297, F1137–F1152. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.; Bonkovsky, H.L. Increased heme oxygenase-1 gene expression in liver cells and splanchnic organs from portal hypertensive rats. Hepatology 1999, 29, 1672–1679. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Schroeder, J.L.; Bammler, T.; Beyer, R.; Srinouanprachanh, S. Gene Expression Profiling of Nephrotoxicity from the Sevoflurane Degradation Product Fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl Ether (“Compound A”) in Rats. Toxicol. Sci. 2005, 90, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.; Afshari, C.A.; Amin, R.P.; Bertram, T.A.; Car, B.; Cunningham, M.; Kind, C.; Kramer, J.A.; Lawton, M.; Mirsky, M.; et al. Identification of platform-independent gene expression markers of cisplatin nephrotoxicity. Environ. Health Perspect. 2004, 112, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Dieterich, C.; Puey, A.; Lyn, S.; Swezey, R.; Furimsky, A.; Fairchild, D.; Mirsalis, J.C.; Ng, H.H. Gene Expression Analysis Reveals New Possible Mechanisms of Vancomycin-Induced Nephrotoxicity and Identifies Gene Markers Candidates. Toxicol. Sci. 2008, 107, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Liguori, M.J.; Sparkenbaugh, E.M.; Waring, J.F.; Blomme, E.A.G.; Ganey, P.E.; Roth, R.A. Gene Expression Profiles in Livers from Diclofenac-Treated Rats Reveal Intestinal Bacteria-Dependent and -Independent Pathways Associated with Liver Injury. J. Pharmacol. Exp. Ther. 2008, 327, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, M.; Fernandez, P.; Guillen, M. Anti-Inflammatory Actions of the Heme Oxygenase-1 Pathway. Curr. Pharm. Des. 2003, 9, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Hill-Kapturczak, N.; Chang, S.-H.; Agarwal, A. Heme Oxygenase and the Kidney. DNA Cell Biol. 2002, 21, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhao, L.-F.; Zhao, Z.-F.; Wang, Y.; Zhao, J.-J.; Zhang, L. Heme oxygenase-1 prevents liver fibrosis in rats by regulating the expression of PPARγ and NF-κB. World J. Gastroenterol. 2012, 18, 1680–1688. [Google Scholar] [CrossRef]
- Morishita, K.; Mizukawa, Y.; Kasahara, T.; Okuyama, M.; Takashima, K.; Toritsuka, N.; Miyagishima, T.; Nagao, T.; Urushidani, T. Gene Expression Profile in Liver of Differing Ages of Rats After Single Oral Administration of Acetaminophen. J. Toxicol. Sci. 2006, 31, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.; Speyer, B.E.; Billing, B.H. Serum haptoglobin in liver disease. Gut 1961, 2, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Robertson, L.D.; Roper, D. Laboratory Methods Used in the Investigation of the Haemolytic Anaemias. In Dacie and Lewis Practical Haematology; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 214–227. [Google Scholar]
- Fagoonee, S.; Gburek, J.; Hirsch, E.; Marro, S.G.; Moestrup, S.K.; Laurberg, J.M.; Christensen, E.I.; Silengo, L.; Altruda, F.; Tolosano, E. Plasma Protein Haptoglobin Modulates Renal Iron Loading. Am. J. Pathol. 2005, 166, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Mercadante, S.; Arcuri, E. Opioids and renal function. J. Pain 2004, 5, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.V.; Howse, M.; Bell, G.; Henry, J. Substance abuse and the kidney. QJM Int. J. Med. 2000, 93, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.P.; Singh, N.; Jaggi, A.S. A Review on Renal Toxicity Profile of Common Abusive Drugs. Korean J. Physiol. Pharmacol. 2013, 17, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alinejad, S.; Ghaemi, K.; Abdollahi, M.; Mehrpour, O. Nephrotoxicity of methadone: A systematic review. SpringerPlus 2016, 5, 2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombi, M.; Muryan, A.; Canzonieri, R.; Trimarchi, H. Biomarcadores en la lesión renal aguda: ¿ Paradigma o evidencia? Nefrología 2016, 36, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasung, M.E.; Chawla, L.S.; Madero, M. Biomarkers of renal function, which and when? Clin. Chim. Acta 2015, 438, 350–357. [Google Scholar] [CrossRef]
- Çuhadar, S. Serum Cystatin C as a Biomarker. In Biomarkers in Kidney Disease; Springer Science and Business Media LLC: Berlin, Germany, 2016; pp. 445–461. [Google Scholar]
- Lezaic, V. Albuminuria as a Biomarker of the Renal Disease; Springer Science and Business Media LLC: Berlin, Germany, 2016; pp. 427–444. [Google Scholar]
- Chugh, S.S.; Macé, C.; Clément, L.; Avila, M.D.N.; Marshall, C.B. Angiopoietin-like 4 based therapeutics for proteinuria and kidney disease. Front. Pharmacol. 2014, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.; Moradi, H. Dual role of circulating angiopoietin-like 4 (ANGPTL4) in promoting hypertriglyceridemia and lowering proteinuria in nephrotic syndrome. Am. J. Kidney Dis. 2014, 64, 495–498. [Google Scholar] [CrossRef] [Green Version]
- Tynkevich, E.; Flamant, M.; Haymann, J.-P.; Metzger, M.; Thervet, E.; Boffa, J.-J.; Vrtovsnik, F.; Houillier, P.; Froissart, M.; Stengel, B.; et al. Urinary creatinine excretion, measured glomerular filtration rate and CKD outcomes. Nephrol. Dial. Transpl. 2015, 30, 1386–1394. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.D.; Rapoport, M.; Cooperband, S.R. The Renal Clearance of Amylase in Renal Insufficiency, Acute Pancreatitis, and Macroamylasemia. Ann. Intern. Med. 1969, 71, 919. [Google Scholar] [CrossRef] [PubMed]
- Schönebeck, J.; Söderberg, M. Serum Amylase in Renal Failure. Scand. J. Urol. Nephrol. 1971, 5, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Warshaw, A.L. Editorial: The kidney and changes in amylase clearance. Gastroenterology 1976, 71, 702–704. [Google Scholar] [CrossRef]
- Trujillo, M.A. Elevated Serum Amylase. In Decision Making in Medicine—An Algorithmic Approach, 3rd ed.; Mushlin, S.B., Greene, H.L., Eds.; Elsevier Inc.: New York, NY, USA, 2010; pp. 228–229. [Google Scholar] [CrossRef]
- Tsianos, E.V.; Dardamanis, M.A.; Elisaf, M.; Vasakos, S.; Siamopoulos, K.C. The value of alpha-amylase and isoamylase determination in chronic renal failure patients. Int. J. Pancreatol. 1994, 15, 105–111. [Google Scholar]
- Rosenblum, J.L.; Raab, B.K.; Alpers, D.H. Hepatobiliary and pancreatic clearance of circulating pancreatic amylase. Am. J. Physiol. Liver Physiol. 1982, 243, G21–G27. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Joffe, S.N.; McIntosh, W.; Brodie, M.J. Amylase activity in human bile. Gut 1979, 20, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Çuhadar, S.; Semerci, T. Renal Biomarkers N-Acetyl-Beta-d-Glucosaminidase (NAG), Endothelin, and Their Application. In Biomarkers in Kidney Disease; Springer Science and Business Media LLC: New York, NY, USA, 2016; pp. 369–396. [Google Scholar]
- Deroee, A.F.; Nezami, B.G.; Mehr, S.E.; Hosseini, R.; Salmasi, A.H.; Talab, S.S.; Jahanzad, I.; Dehpour, A.R. Cholestasis induced nephrotoxicity: The role of endogenous opioids. Life Sci. 2010, 86, 488–492. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, Y.-H.; Lee, S.-G.; Kang, E.S.; Cha, B.; Kim, J.-H.; Lee, B.W. Urinary N-acetyl-β-D-glucosaminidase, an early marker of diabetic kidney disease, might reflect glucose excursion in patients with type 2 diabetes. Medicine 2016, 95, e4114. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, Y.-H.; Lee, S.-G.; Kang, E.S.; Cha, B.-S.; Lee, B.-W. The renal tubular damage marker urinary N-acetyl-β-D-glucosaminidase may be more closely associated with early detection of atherosclerosis than the glomerular damage marker albuminuria in patients with type 2 diabetes. Cardiovasc. Diabetol. 2017, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Udomah, F.P.; Ekrikpo, U.; Effa, E.; Salako, B.; Arije, A.; Kadiri, S. Association between Urinary N-Acetyl-Beta-D-Glucosaminidase and Microalbuminuria in Diabetic Black Africans. Int. J. Nephrol. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Asaka, M.; Nagase, K.; Miyazaki, T.; Alpert, E. Aldolase A Isoenzyme Levels in Serum and Tissues of Patients with Liver Diseases. Gastroenterology 1983, 84, 155–160. [Google Scholar] [CrossRef]
- Bell, L.N.; Vuppalanchi, R.; Watkins, P.B.; Bonkovsky, H.L.; Serrano, J.; Fontana, R.J.; Wang, M.; Rochon, J.; Chalasani, N.; US Drug-Induced Liver Injury Network (DILIN) Research Group. Serum proteomic profiling in patients with drug-induced liver injury. Aliment. Pharmacol. Ther. 2012, 35, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, L.E.; Skinner, C.M.; Quick, C.M.; Kennon-McGill, S.; McGill, M.R.; Walker, L.A.; ElSohly, M.A.; Gurley, B.J.; Koturbash, I. Hepatotoxicity of a Cannabidiol-Rich Cannabis Extract in the Mouse Model. Molecules 2019, 24, 1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehinto, A.C.; Prucha, M.S.; Colli-Dula, R.C.; Kroll, K.J.; Lavelle, C.M.; Barber, D.S.; Vulpe, C.; Denslow, N.D. Gene networks and toxicity pathways induced by acute cadmium exposure in adult largemouth bass (Micropterus salmoides). Aquat. Toxicol. 2014, 152, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Soontrapa, K.; Ito, Y.; Iwaisako, K.; Moniaga, C.S.; Asagiri, M.; Majima, M.; Narumiya, S. Hepatic stellate cells relay inflammation signaling from sinusoids to parenchyma in mouse models of immune-mediated hepatitis. Hepatology 2015, 63, 1325–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidek, N.; Hellmann, J.; Kramer, P.-J.; Hewitt, P. Acute Hepatotoxicity: A Predictive Model Based on Focused Illumina Microarrays. Toxicol. Sci. 2007, 99, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, G.; Calcagno, G.; Sibillo, R.; Cuomo, R.; Nardone, G.; Castellano, L.; Blanco, C.D.V.; Budillon, G.; Salvatore, F. Quantitative Analysis of Aldolase A mRNA in Liver Discriminates between Hepatocellular Carcinoma and Cirrhosis. Clin. Chem. 2000, 46, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, P.; Liang, B.; He, S.; Tan, W.; Zhang, X.; Su, C.; Zhao, L.; Wei, L.; Chen, Y.; et al. Long-chain fatty acid activates hepatocytes through CD36 mediated oxidative stress. Lipids Health Dis. 2018, 17, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Chen, Y.; Ma, Y.; Liu, T.; Zhao, M.; Wang, Z.; Zhao, L. Lipidomic Profiling Reveals Disruption of Lipid Metabolism in Valproic Acid-Induced Hepatotoxicity. Front. Pharmacol. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, V.; Arivalagan, S.; Siddique, A.I.; Namasivayam, N. Antihyperlipidemic and antiapoptotic potential of zingerone on alcohol induced hepatotoxicity in experimental rats. Chem. Interact. 2017, 272, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: Structure, function, regulation, and role in disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Sheriff, S.A.; Devaki, T.; Chandy, S. Lycopene stabilizes lipoprotein levels during D-galactosamine/lipopolysaccharide induced hepatitis in experimental rats. Asian Pac. J. Trop. Biomed. 2012, 2, 975–980. [Google Scholar] [CrossRef] [Green Version]
- La Paglia, L.; Listì, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhang, R.; Yang, S.; Chen, F.; Shi, J. Knockdown of ANGPTL-4 inhibits inflammatory response and extracellular matrix accumulation in glomerular mesangial cells cultured under high glucose condition. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3368–3373. [Google Scholar] [CrossRef] [Green Version]
- Al Shawaf, E.; Abu-Farha, M.; Devarajan, S.; Alsairafi, Z.; Al-Khairi, I.; Cherian, P.; Ali, H.; Mathur, A.; Al-Mulla, F.; Al Attar, A.; et al. ANGPTL4: A Predictive Marker for Diabetic Nephropathy. J. Diabetes Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Del Nogal-Avila, M.; Donoro-Blazquez, H.; Saha, M.K.; Marshall, C.B.; Clement, L.C.; Macé, C.E.A.; Chugh, S.S. Novel therapeutic approaches for chronic kidney disease due to glomerular disorders. Am. J. Physiol. Renal Physiol. 2016, 311, F63–F65. [Google Scholar] [CrossRef]
- Koliwad, S.K.; Gray, N.E.; Wang, J.-C. Angiopoietin-like 4 (Angptl4). Adipocyte 2012, 1, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2013, 55, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukonina, V.; Lookene, A.; Olivecrona, T.; Olivecrona, G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, F.; Rodriguez-Soriano, J.; Prieto, J.A.; Elorz, J.; Aguirre, M.; Ariceta, G.; Martin, S.; Sanjurjo, P.; Aldámiz-Echevarría, L.; Rodr, J.; et al. The Arginine-Creatine Pathway is Disturbed in Children and Adolescents with Renal Transplants. Pediatr. Res. 2008, 64, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, R.P.; Vickers, A.E.; Sistare, F.; Thompson, K.L.; Roman, R.J.; Lawton, M.; Kramer, J.; Hamadeh, H.K.; Collins, J.; Grissom, S.; et al. Identification of putative gene based markers of renal toxicity. Environ. Health Perspect. 2004, 112, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; Moats, J.C.; Harris, R.M.; Osna, N.A.; Thomes, P.G.; Tuma, D.J. Alcohol Consumption Decreases Rat Hepatic Creatine Biosynthesis Via Altered Guanidinoacetate Methyltransferase Activity. Alcohol. Clin. Exp. Res. 2013, 38, 641–648. [Google Scholar] [CrossRef]
- Mollet, G.; Ratelade, J.; Boyer, O.; Muda, A.O.; Morisset, L.; Lavin, T.A.; Kitzis, D.; Dallman, M.; Bugeon, L.; Hubner, N.; et al. Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J. Am. Soc. Nephrol. 2009, 20, 2181–2189. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaeifar, M.; Wlodkowski, T.; Simic, I.; Denc, H.; Mollet, G.; Weber, S.; Moyers, J.J.; Brühl, B.; Randles, M.J.; Lennon, R.; et al. An inducible mouse model of podocin-mutation-related nephrotic syndrome. PLoS ONE 2017, 12, e0186574. [Google Scholar] [CrossRef]
- Oleggini, R.; Bertelli, R.; Di Donato, A.; Di Duca, M.; Caridi, G.; Sanna, S.-C.; Scolari, F.; Murer, L.; Allegri, L.; Coppo, R.; et al. Rare Functional Variants of Podocin (NPHS2) Promoter in Patients with Nephrotic Syndrome. Gene Expr. 2006, 13, 59–66. [Google Scholar] [CrossRef]
- Roselli, S.; Heidet, L.; Sich, M.; Henger, A.; Kretzler, M.; Gubler, M.-C.; Antignac, C. Early Glomerular Filtration Defect and Severe Renal Disease in Podocin-Deficient Mice. Mol. Cell. Biol. 2004, 24, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Xiong, C.; Wu, Q.; Fang, M.; Li, H.; Chen, B.; Chi, T. Protective effects of luteolin on nephrotoxicity induced by long-term hyperglycaemia in rats. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.-W.; Nissaisorakarn, P.; Husain, I.; Jim, B. Proteinuric Kidney Diseases: A Podocyte’s Slit Diaphragm and Cytoskeleton Approach. Front. Med. 2018, 5, 221. [Google Scholar] [CrossRef]
- Di Duca, M.; Oleggini, R.; Sanna-Cherchi, S.; Pasquali, L.; Di Donato, A.; Parodi, S.; Bertelli, R.; Caridi, G.; Frasca, G.; Cerullo, G.; et al. Cis and trans regulatory elements in NPHS2 promoter: Implications in proteinuria and progression of renal diseases. Kidney Int. 2006, 70, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, C.; Crane, J. Tramadol deaths in Northern Ireland: A review of cases from 1996 to 2012. J. Forensic Leg. Med. 2014, 23, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.E.; Lacy, J.M.; Fligner, C.L.; Thiersch, N.; Howard, J.; Harruff, R.C.; Logan, B.K. Tramadol (Ultram) concentrations in death investigation and impaired driving cases and their significance. J. Forensic Sci. 2004, 49, 1–5. [Google Scholar] [CrossRef]
- Mannocchi, G.; Napoleoni, F.; Napoletano, S.; Pantano, F.; Santoni, M.; Tittarelli, R.; Arbarello, P. Fatal self administration of tramadol and propofol: A case report. J. Forensic Leg. Med. 2013, 20, 715–719. [Google Scholar] [CrossRef]
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Çelik, H.; Karaoğlanoğlu, M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 2006, 27, 339–344. [Google Scholar] [CrossRef]
- Slott, P.A.; Liu, M.H.; Tavoloni, N. Origin, pattern, and mechanism of bile duct proliferation following biliary obstruction in the rat. Gastroenterology 1990, 99, 466–477. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 2018, 69, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2007, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Matthiesen, T.; Wöhrmann, T.; Coogan, T.P.; Uragg, H. The experimental toxicology of tramadol: An overview. Toxicol. Lett. 1998, 95, 63–71. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Christoph, T.; Kögel, B.; Schiene, K.; Hennies, H.-H.; Englberger, W.; Haurand, M.; Jahnel, U.; Cremers, T.I.F.H.; Friderichs, E.; et al. (–)-(1R,2R)-3-(3-Dimethylamino-1-ethyl-2-methyl-propyl)-phenol Hydrochloride (Tapentadol HCl): A Novel μ-Opioid Receptor Agonist/Norepinephrine Reuptake Inhibitor with Broad-Spectrum Analgesic Properties. J. Pharmacol. Exp. Ther. 2007, 323, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Williams, J.A.; Stadtman, E.P.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994, 233, 346–357. [Google Scholar] [CrossRef]
- Costa, I.; Carvalho, F.; Magalhães, T.; Pinho, P.G.; Silvestre, R.; Dinis-Oliveira, R.J. Promising blood-derived biomarkers for estimation of the postmortem interval. Toxicol. Res. 2015, 4, 1443–1452. [Google Scholar] [CrossRef] [Green Version]
- Yalamati, P.; Bhongir, A.V.; Karra, M.; Beedu, S.R. Comparative Analysis of Urinary Total Proteins by Bicinchoninic Acid and Pyrogallol Red Molybdate Methods. J. Clin. Diagn. Res. 2015, 9, BC01–BC04. [Google Scholar] [CrossRef]
- Gong, H.; Wang, X.; Wei, J.; Pan, L.; Shi, Y.; Lin, H. The Role of CD36 in the Effect of Arginine in Atherosclerotic Rats. Med. Sci. Monit. 2015, 21, 1494–1499. [Google Scholar] [CrossRef]
- Aliparasti, M.R.; Alipour, M.R.; Almasi, S.; Feizi, H. Effect of Ghrelin on Aldolase Gene Expression in the Heart of Chronic Hypoxic Rat. Int. J. Endocrinol. Metab. 2012, 10, 553–557. [Google Scholar] [CrossRef]
- Nishimura, J.; Dewa, Y.; Okamura, T.; Jin, M.; Saegusa, Y.; Kawai, M.; Umemura, T.; Shibutani, M.; Mitsumori, K. Role of Nrf2 and Oxidative stress on Fenofibrate-Induced Hepatocarcinogenesis in Rats. Toxicol. Sci. 2008, 106, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Mello, T.; Nakatsuka, A.; Fears, S.; Davis, W.; Tsukamoto, H.; Bosron, W.F.; Sanghani, S.P. Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun. 2008, 374, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Chen, H.-L.; Sun, S.S.; Li, H.L.; Zhang, J.W.; Cao, L. Effect of Angiopoietin-Like Protein 4 on Severe Acute Pancreatitis-induced Lung Injury in Rats. J. Clin. Cell. Immunol. 2013, 4, 4. [Google Scholar] [CrossRef]
- Yamashita, Y.; Ueyama, T.; Nishi, T.; Yamamoto, Y.; Kawakoshi, A.; Sunami, S.; Iguchi, M.; Tamai, H.; Ueda, K.; Ito, T.; et al. Nrf2-Inducing Anti-Oxidation Stress Response in the Rat Liver—New Beneficial Effect of Lansoprazole. PLoS ONE 2014, 9, e97419. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-Y.; Chen, L.-Z.; Jiang, X.-Y.; Mo, Y.; Ling, Y.-H.; Sun, L.-Z. Temporal and Spatial Expression of Podocyte-Associated Molecules Are Accompanied by Proteinuria in IgA Nephropathy Rat Model. Physiol. Res. 2013, 62, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.P.; Clow, K.; Brosnan, J.T.; Brosnan, M.E. Synthesis of guanidinoacetate and creatine from amino acids by rat pancreas. Br. J. Nutr. 2013, 111, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, R.; Zhu, C.; Ding, G.; Zhao, M.; Bai, M.; Jia, Z.; Zhang, A.; Huang, S. Huaier Cream Protects against Adriamycin-Induced Nephropathy by Restoring Mitochondrial Function via PGC-1α Upregulation. PPAR Res. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J.; Duarte, J.A.; Remiao, F.; Navarro, A.S.; Bastos, M.D.L.; Carvalho, F. Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 2006, 227, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J.; Remiao, F.; Duarte, J.A.; Ferreira, R.; Navarro, A.S.; Bastos, M.D.L.; Carvalho, F. P-glycoprotein induction: An antidotal pathway for paraquat-induced lung toxicity. Free. Radic. Biol. Med. 2006, 41, 1213–1224. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | No. of Amplification Cycles | Reference |
|---|---|---|---|---|
| Cd36 (Cluster of differentiation 36/fatty acid translocase) | AGGAAGTGGCAAAGAATAGCAG | ACAGACAGTGAAGGCTCAAAGA | 37 | [233] |
| Aldoa (Fructose-bisphosphate aldolase A) | ATGCCCCACCCATACCCAGCACT | AGCAGCAGTTGGCGGTAGAAGCG | 37 | [234] |
| Apex1 (Apurinic/apyrimidinic endonuclease 1) | GAATGTGGATGGGCTTCGA | AAGATGTCTGGTGCTTCTTCCTTT | 41 | [235] |
| Lpl (Lipoprotein lipase) | CTTAAGTGGAAGAACGACTCCTACT | GTCATGGCATTTCACAAACACTGCA | 41 | [236] |
| Angptl4 (Angiopoietin-like 4) | GCCGCTACTATCCACTAC | CCTGTTGCTCTGACTGTT | 45 | [237] |
| Hmox1 (Heme oxygenase 1) | ACAGGGTGACAGAAGAGGCTAA | CTGTGAGGGACTCTGGTCTTTG | 45 | [238] |
| Nphs2 (Podocin) | TGGAAGCTGAGGCACAAAGA | AGAATCTCAGCCGCCATCCT | 38 | [239] |
| Gamt (Guanidinoacetate N-methyltransferase) | ACTCATGCTTTCCGTTTGCT | AGGCACCTGAGTCTCCTCAA | 38 | [240] |
| 18S rRNA (18S ribosomal RNA) | TTCGGAACTGAGGCCATGATT | TTTCGCTCTGGTCCGTCTTG | In line with that of the target gene | [241] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, J.; Faria, J.; Garcez, F.; Leal, S.; Afonso, L.P.; Nascimento, A.V.; Moreira, R.; Queirós, O.; Carvalho, F.; Dinis-Oliveira, R.J. Repeated Administration of Clinical Doses of Tramadol and Tapentadol Causes Hepato- and Nephrotoxic Effects in Wistar Rats. Pharmaceuticals 2020, 13, 149. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070149
Barbosa J, Faria J, Garcez F, Leal S, Afonso LP, Nascimento AV, Moreira R, Queirós O, Carvalho F, Dinis-Oliveira RJ. Repeated Administration of Clinical Doses of Tramadol and Tapentadol Causes Hepato- and Nephrotoxic Effects in Wistar Rats. Pharmaceuticals. 2020; 13(7):149. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070149
Chicago/Turabian StyleBarbosa, Joana, Juliana Faria, Fernanda Garcez, Sandra Leal, Luís Pedro Afonso, Ana Vanessa Nascimento, Roxana Moreira, Odília Queirós, Félix Carvalho, and Ricardo Jorge Dinis-Oliveira. 2020. "Repeated Administration of Clinical Doses of Tramadol and Tapentadol Causes Hepato- and Nephrotoxic Effects in Wistar Rats" Pharmaceuticals 13, no. 7: 149. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13070149