
New Uncharged 2-Thienostilbene Oximes as Reactivators of Organophosphate-Inhibited Cholinesterases

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
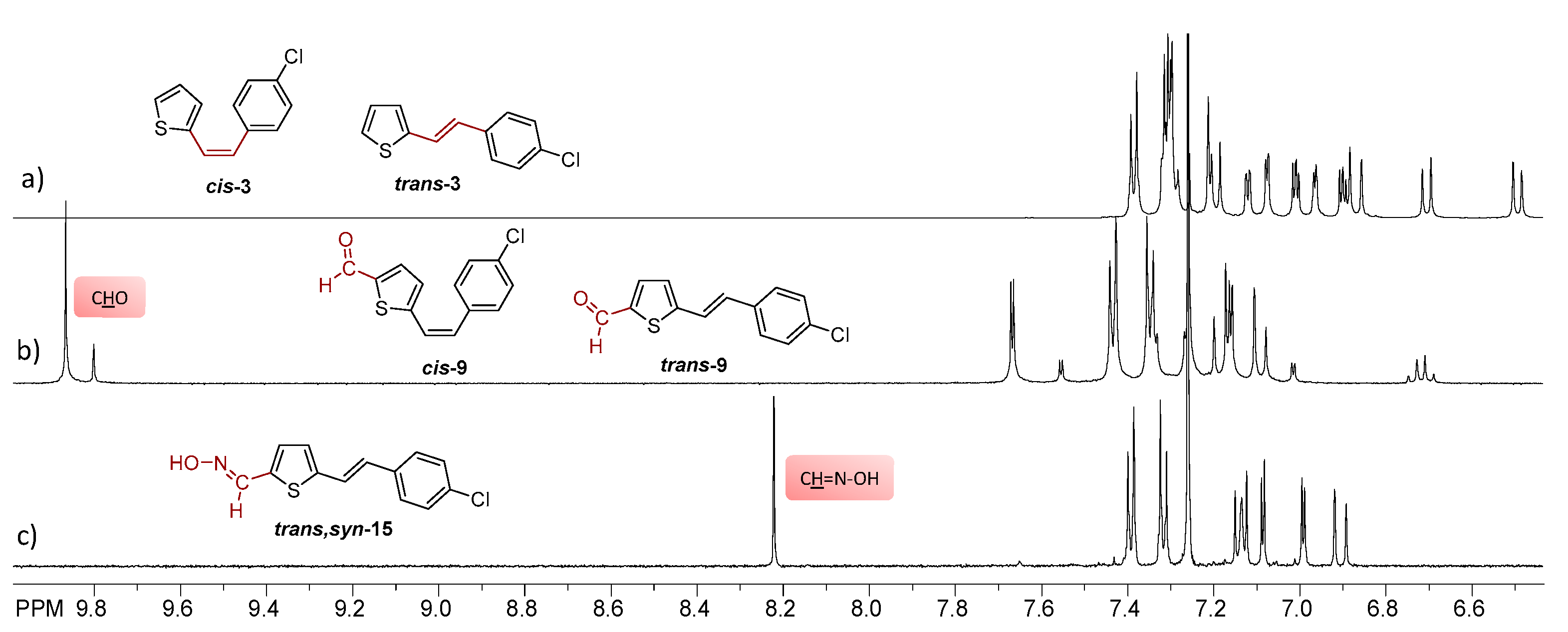
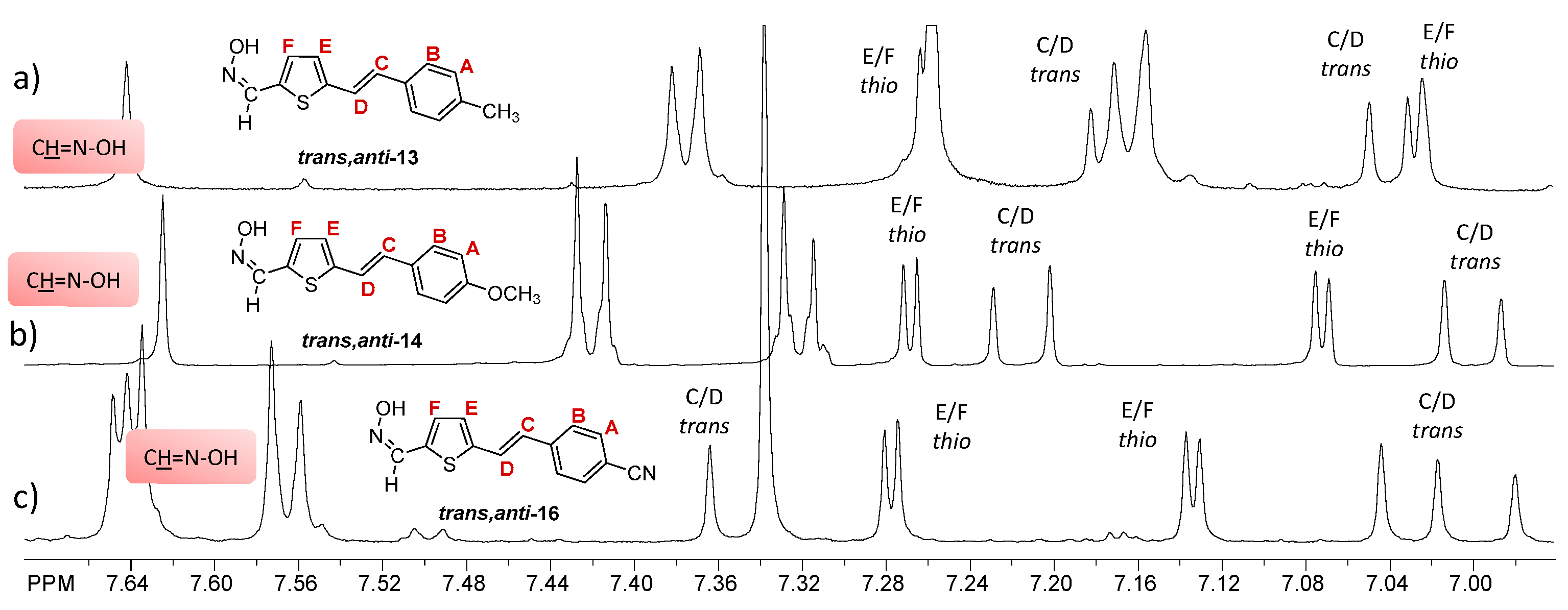
2.1. Synthesis and Spectroscopic Characterization of New Thienostilbene Oximes 13–18
2.2. In Silico Prediction of ADME Properties of Thienostilbene Oximes 13–18
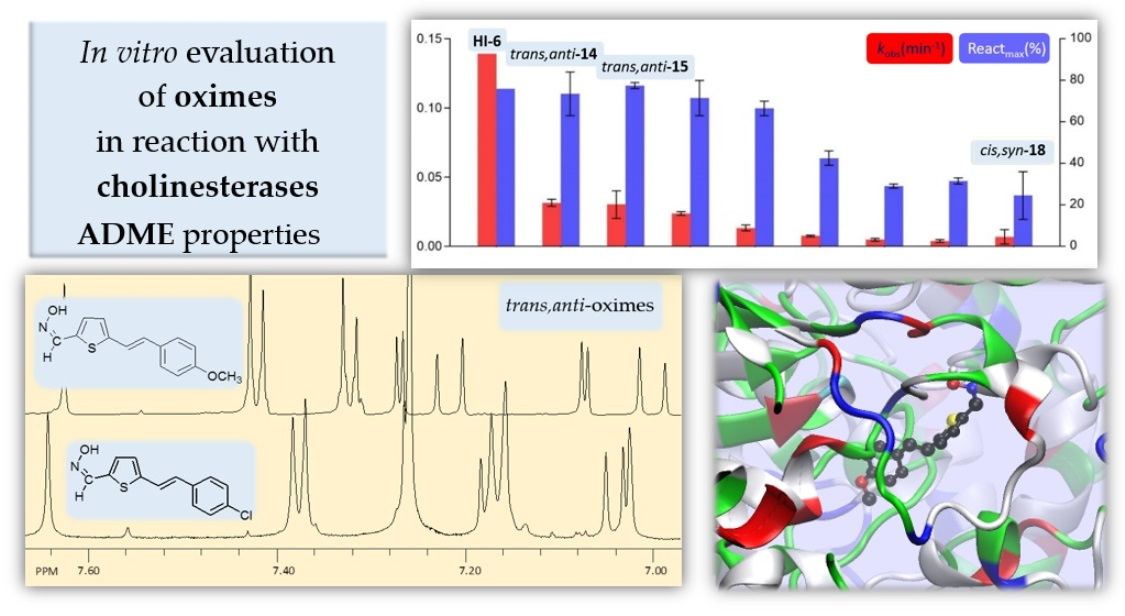
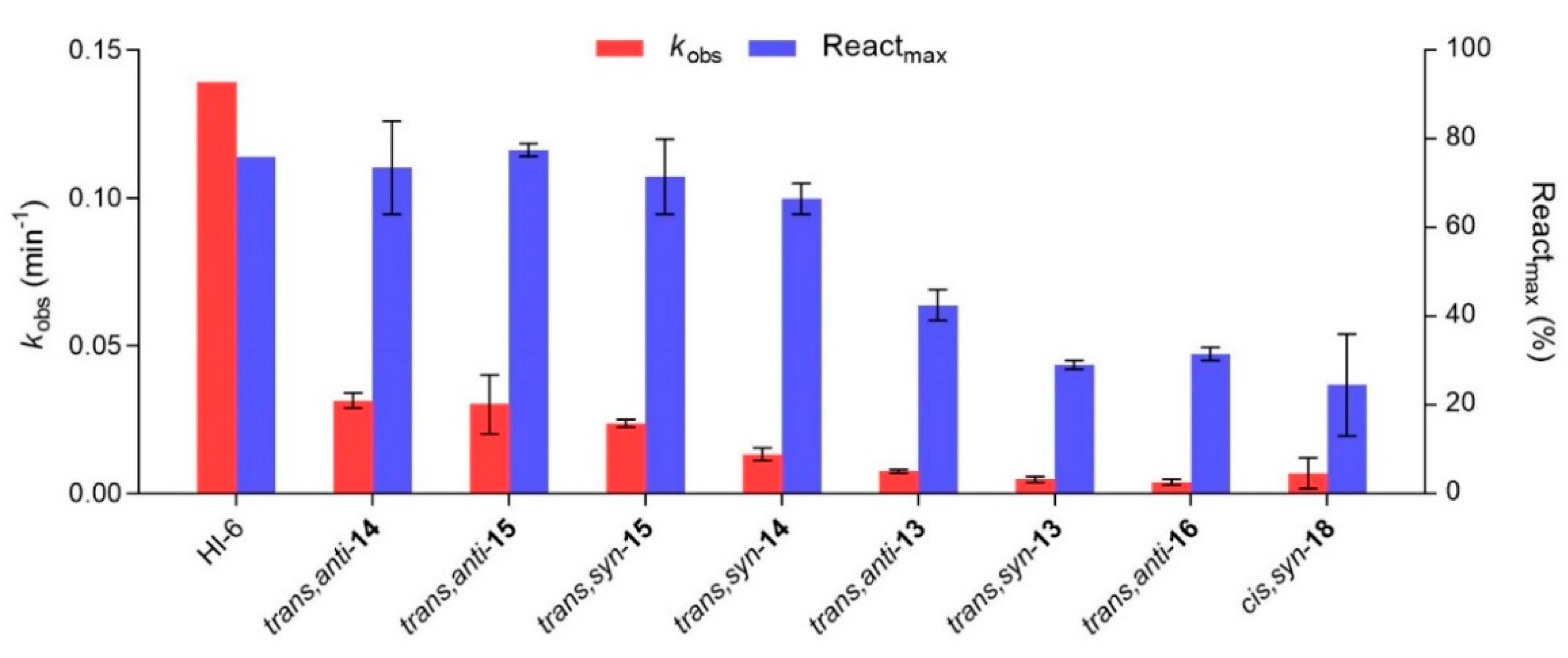
2.3. In Vitro Evaluation of Thienostilbene Oximes 13–18 in Reactions with Cholinesterases
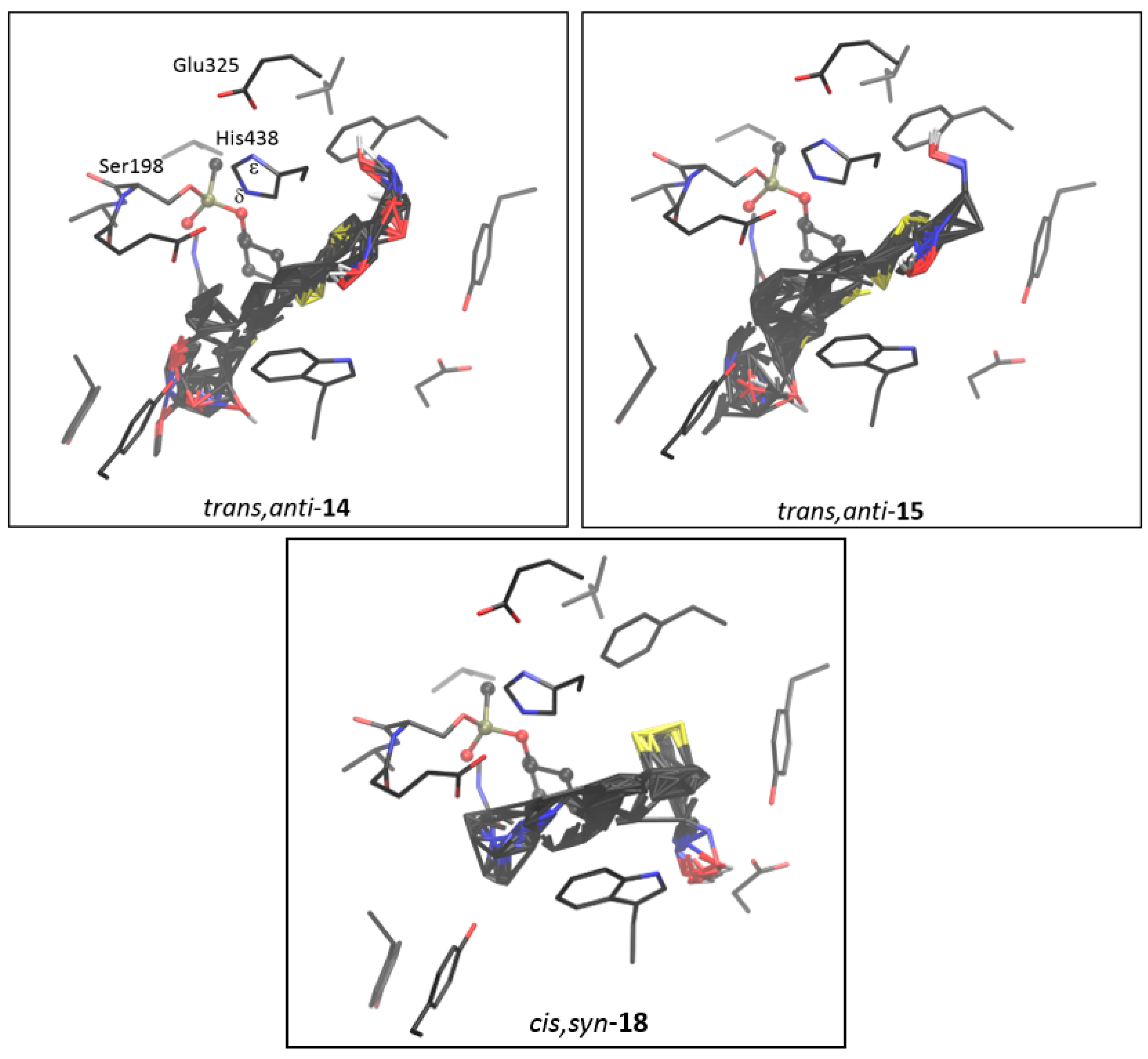
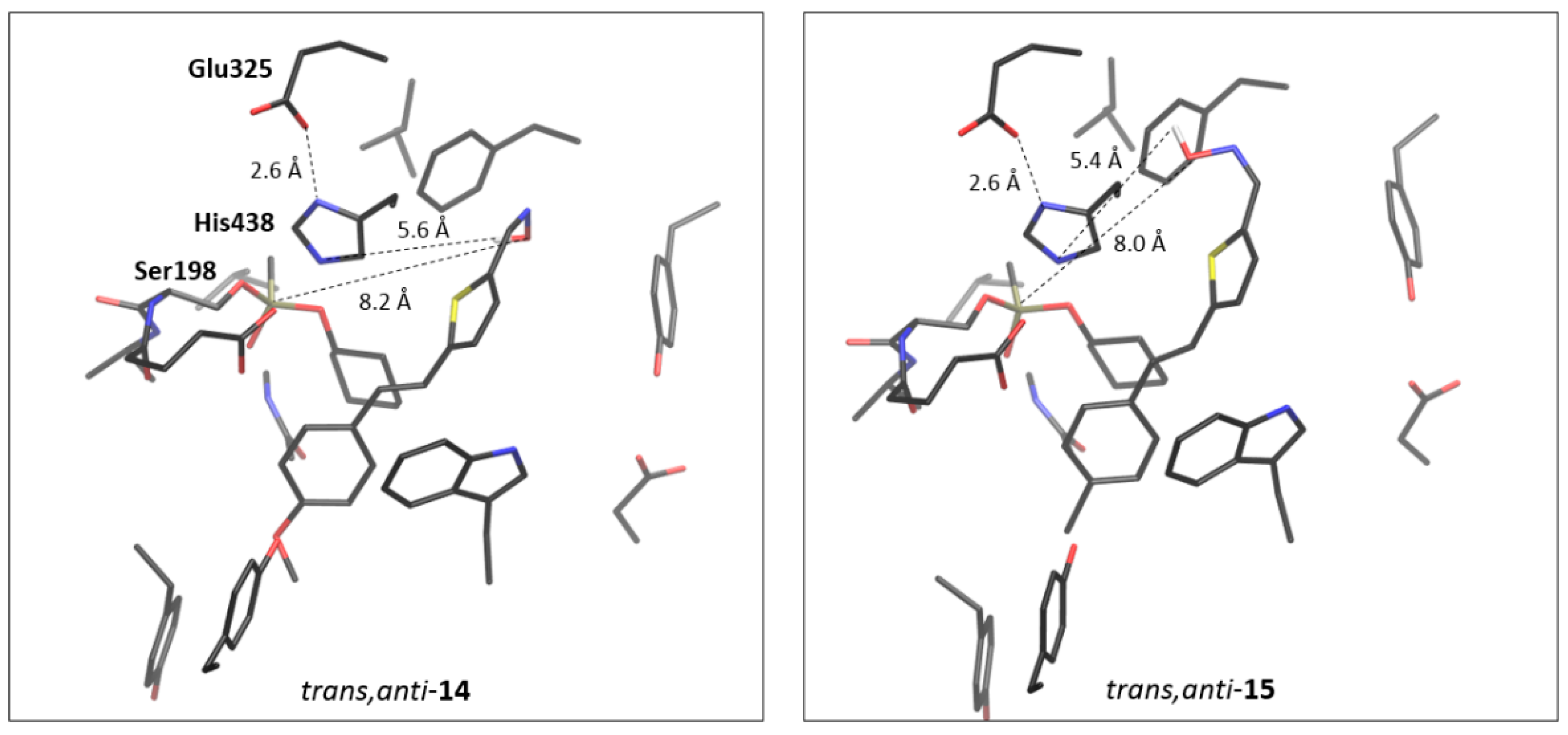
2.4. Docking of Potential Reactivators
3. Materials and Methods
3.1. General
3.2. Synthesis of Thienostilbenes 1–6






3.3. Synthesis of Aldehydes 7–12






3.4. Synthesis of Oximes 13–18






3.5. Chemicals for Enzyme Assays
3.6. Cholinesterase Activity Measurements
3.7. Docking of Thienostilbene Oximes
3.8. In Silico Prediction of ADME Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dolgin, E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med. 2013, 19, 1194. [Google Scholar] [CrossRef]
- Steindl, D.; Boehmerle, W.; Körner, R.; Praeger, D.; Haug, M.; Nee, J.; Schreiber, A.; Scheibe, F.; Demin, K.; Jacoby, P.; et al. Novichok nerve agent poisoning. Lancet 2021, 397, 249–252. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, A.D.; Geula, C. Neurobiology of Butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Butyrylcholinesterase: Its Function and Inhibitors, 1st ed.; Martin, D., Ed.; Taylor & Francis Group plc: London, UK, 2003; ISBN 1841842095. [Google Scholar]
- Čadež, T.; Kovarik, Z. Advancements in recombinant technology for production of butyrylcholinesterase, a bioscavenger of nerve agent. Period. Biol. 2020, 121–122, 55–63. [Google Scholar] [CrossRef]
- Sidell, F.R.; Borak, J. Chemical warfare agents: II. nerve agents. Ann. Emerg. Med. 1992, 21, 865–871. [Google Scholar] [CrossRef]
- Timperley, C.M.; Abdollahi, M.; Al-Amri, A.S.; Baulig, A.; Benachour, D.; Borrett, V.; Cariño, F.A.; Geist, M.; Gonzalez, D.; Kane, W.; et al. Advice on assistance and protection by the Scientific Advisory Board of the Organisation for the Prohibition of Chemical Weapons: Part 2. On preventing and treating health effects from acute, prolonged, and repeated nerve agent exposure, and the identific. Toxicology 2019, 413, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timperley, C.M.; Forman, J.E.; Abdollahi, M.; Al-Amri, A.S.; Baulig, A.; Benachour, D.; Borrett, V.; Cariño, F.A.; Geist, M.; Gonzalez, D.; et al. Advice on assistance and protection provided by the Scientific Advisory Board of the Organisation for the Prohibition of Chemical Weapons: Part 1. On medical care and treatment of injuries from nerve agents. Toxicology 2019, 415, 56–69. [Google Scholar] [CrossRef]
- Maček Hrvat, N.; Kovarik, Z. Counteracting poisoning with chemical warfare nerve agents. Arh. Hig. Rada Toksikol. 2021, 71, 266–284. [Google Scholar] [CrossRef] [PubMed]
- Zorbaz, T.; Kovarik, Z. Neuropharmacology: Oxime antidotes for organophosphate pesticide and nerve agent poisoning. Period. Biol. 2020, 121–122, 35–54. [Google Scholar] [CrossRef]
- Zorbaz, T.; Mišetić, P.; Probst, N.; Žunec, S.; Zandona, A.; Mendaš, G.; Micek, V.; MačEk Hrvat, N.; Katalinić, M.; Braïki, A.; et al. Pharmacokinetic evaluation of brain penetrating morpholine-3-hydroxy-2-pyridine oxime as an antidote for nerve agent poisoning. ACS Chem. Neurosci. 2020, 11, 1072–1084. [Google Scholar] [CrossRef]
- Sit, R.K.; Kovarik, Z.; Maček Hrvat, N.; Žunec, S.; Green, C.; Fokin, V.V.; Sharpless, K.B.; Radic, Z.; Taylor, P. Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther. 2018, 367, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Jun, D.; Musilek, K. Structural requirements of acetylcholinesterase reactivators. Mini Rev. Med. Chem. 2006, 6, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Norrrahim, M.N.F.; Ahmad Shah, N.A.; Jamal, S.H.; Yunus, W.M.Z.W.; Ernest, V.F.K.V.; Kasim, N.A.M. Nanocellulose-based filters as novel barrier systems for chemical warfare agents. Solid State Phenom. 2021, 317, 180–186. [Google Scholar] [CrossRef]
- de Koning, M.C.; van Grol, M.; Noort, D. Peripheral site ligand conjugation to a non-quaternary oxime enhances reactivation of nerve agent-inhibited human acetylcholinesterase. Toxicol. Lett. 2011, 206, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Kliachyna, M.; Santoni, G.; Nussbaum, V.; Renou, J.; Sanson, B.; Colletier, J.P.; Arboléas, M.; Loiodice, M.; Weik, M.; Jean, L.; et al. Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- Aldoxime and -Amidoxime hybrids as efficient uncharged reactivators of nerve agent-Inhibited human acetylcholinesterase. Eur. J. Med. Chem. 2014, 78, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Šagud, I.; Škorić, I.; Šindler-Kulyk, M. Excited state transformations of heterostilbenes: Pathways to polycyclic skeleta. Comptes Rendus. Chim. 2018, 21, 1043–1052. [Google Scholar] [CrossRef]
- Ribeiro, T.S.; Prates, A.; Alves, S.R.; Oliveira-Silva, J.J.; Riehl, C.A.S.; Figueroa-Villar, J.D. The effect of neutral oximes on the reactivation of human acetylcholinesterase inhibited with paraoxon. J. Braz. Chem. Soc. 2012, 23, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Porcheddu, A.; Giacomelli, G. Synthesis of Oximes and Hydroxamic Acids. In The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids; Liebman, J.F., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 2009; pp. 163–231. [Google Scholar]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRX 2005, 2, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Testa, B.; Crivori, P.; Reist, M.; Carrupt, P.A. The influence of lipophylicity on the pharmacokinetic behavior of drugs: Concepts and examples. Perspect. Drug Discov. Des. 2000, 19, 179–211. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocks, M. The small molecule drug discovery process-from target selection to candidate selection. In Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies; Jefferis, R., Roberts, S., Ganellin, R., Eds.; The University of Nottingham: England, UK, 2013; pp. 81–126. [Google Scholar] [CrossRef]
- Zandona, A.; Katalinić, M.; Šinko, G.; Radman Kastelic, A.; Primožič, I.; Kovarik, Z. Targeting organophosphorus compounds poisoning by novel quinuclidine-3 oximes: Development of butyrylcholinesterase-based bioscavengers. Arch. Toxicol. 2020, 94, 3157–3171. [Google Scholar] [CrossRef]
- Artursson, E.; Andersson, P.O.; Akfur, C.; Linusson, A.; Borjegren, S.; Ekstrom, F. Catalytic-site conformational equilibrium in nerve-agent adducts of acetylcholinesterase; possible implications for the HI-6 antidote substrate specificity. Biochem. Pharmacol. 2013, 85, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Cuya, T.; da Silva Gonçalves, A.; da Silva, J.A.V.; Ramalho, T.C.; Kuca, C.C.K.; França, T.C.C. The role of the oximes HI-6 and HS-6 inside human acetylcholinesterase inhibited with nerve agents: A computational study. J. Biomol. Struct. Dyn. 2018, 36, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Maraković, N.; Knežević, A.; Rončević, I.; Brazzolotto, X.; Kovarik, Z.; Šinko, G. Enantioseparation, in vitro testing, and structural characterization of triple-binding reactivators of organophosphate-inhibited cholinesterases. Biochem. J. 2020, 477, 2771–2790. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K.; Singh, R.; Singh, K.N. Decarboxylative arylation of α,β-unsaturated carboxylic acids using aryl trazenes by copper/ionic liquid combination in PEG-400. Eur. J. Org. Chem. 2018, 2018, 5942–5949. [Google Scholar] [CrossRef]
- Qu, J.; Cao, C.T.; Cao, C. Determining the excited-state substituent constants of furyl and thienyl groups. J. Phys. Org. Chem. 2018, 31, 3799. [Google Scholar] [CrossRef]
- Lee, S.K.; Min, H.Y.; Huh, K.S.; Kim, E.Y.; Lee, E.; Song, S.; Kim, S. Styrylheterocycles: A novel class of inhibitors on lipopolysaccharide-induced nitric oxide production. Bioorganic Med. Chem. Lett. 2003, 13, 3689–3692. [Google Scholar] [CrossRef]
- Jin, J.; Li, X.; Zhang, J.; Zhao, P.; Tian, H. Rational design of double-check mercury ion chemosensors based on photochromic compounds. Isr. J. Chem. 2013, 53, 288–293. [Google Scholar] [CrossRef]
- Jones, G.; Stanforth, S.P. The Vilsmeier reaction of fully conjugated carbocycles and heterocycles. Org. React. 1997, 49, 1–330. [Google Scholar]
- Yoshimura, S.; Takahashi, S.; Kawamata, A.; Kikugawa, K.; Suehiro, H.; Aoki, A. Synthesis of α-substituted alkanoic acids and inhibition of platelet aggregation. Chem. Pharm. Bull. 1978, 26, 685–702. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Yang, X.; Zhao, C.; Jin, K.; Sun, L. Electrogenerated chemiluminiscence of a series of donor-acceptor molecules and X-ray crystallographic evidence for the reaction mechanisms. J. Phys. Chem. C 2007, 111, 9595–9602. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, J.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Kovarik, Z.; Ciban, N.; Radić, Z.; Simeon-Rudolf, V.; Taylor, P. Active site mutant acetylcholinesterase interactions with 2-PAM, HI-6, and DDVP. Biochem. Biophys. Res. Commun. 2006, 342, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Maček Hrvat, N.; Zorbaz, T.; Šinko, G.; Kovarik, Z. The estimation of oxime efficiency is affected by the experimental design of phosphylated acetylcholinesterase reactivation. Toxicol. Lett. 2018, 293, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Li, H.; Li, B.; Ekstrom, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; et al. Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | logP a | Solubility b [mg/mL] | Permeability c [10−6 cm/s] | PPB d | CNS e |
|---|---|---|---|---|---|
| 13 | 3.95 | 0.004 | 23.6 | 83% | −1.86 |
| 14 | 3.70 | 0.009 | 23.7 | 77% | −1.77 |
| 15 | 4.27 | 0.002 | 23.3 | 90% | −2.05 |
| 16 | 3.32 | 0.009 | 23.4 | 77% | −1.82 |
| 17 | 3.58 | 0.01 | 23.6 | 81% | −1.92 |
| 18 | 3.94 | 0.01 | 23.6 | 83% | −1.86 |
| Thienostilbene Oximes | AChE | BChE |
|---|---|---|
| trans,syn-13 | 46 ± 7 | 355 ± 26 |
| trans,anti-13 | 41 ± 4 | 322 ± 38 |
| trans,syn-14 | 67 ± 12 | 271 ± 24 |
| trans,anti-14 | 54 ± 7 | 172 ± 14 |
| trans,syn-15 | 52 ± 12 | 573 ± 35 |
| trans,anti-15 | 76 ± 21 | 482 ± 45 |
| trans,anti-16 | 158 ± 40 | 44 ± 9 |
| cis,syn-18 | 39 ± 11 | 89 ± 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakić, M.; Čadež, T.; Barić, D.; Puček, I.; Ratković, A.; Marinić, Ž.; Lasić, K.; Kovarik, Z.; Škorić, I. New Uncharged 2-Thienostilbene Oximes as Reactivators of Organophosphate-Inhibited Cholinesterases. Pharmaceuticals 2021, 14, 1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111147
Mlakić M, Čadež T, Barić D, Puček I, Ratković A, Marinić Ž, Lasić K, Kovarik Z, Škorić I. New Uncharged 2-Thienostilbene Oximes as Reactivators of Organophosphate-Inhibited Cholinesterases. Pharmaceuticals. 2021; 14(11):1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111147
Chicago/Turabian StyleMlakić, Milena, Tena Čadež, Danijela Barić, Ivana Puček, Ana Ratković, Željko Marinić, Kornelija Lasić, Zrinka Kovarik, and Irena Škorić. 2021. "New Uncharged 2-Thienostilbene Oximes as Reactivators of Organophosphate-Inhibited Cholinesterases" Pharmaceuticals 14, no. 11: 1147. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111147