
Type and Extent of Information on (Potentially Critical) Quality Attributes Described in European Public Assessment Reports for Adalimumab Biosimilars

 ,
,
Abstract
:Highlights
- Comparing adalimumab biosimilars at the level of quality attributes (QAs), as reported in EPARs, showed that the reporting frequencies of QAs vary between biosimilars compared with the same reference biological (Humira®).
- Regulators emphasized reporting of potentially critical QAs (pCQAs) in EPARs and more consistently reported functional pCQAs because they are directly related to the drug mechanisms of action and provide valuable information for clinical performance and the extrapolation of indications.
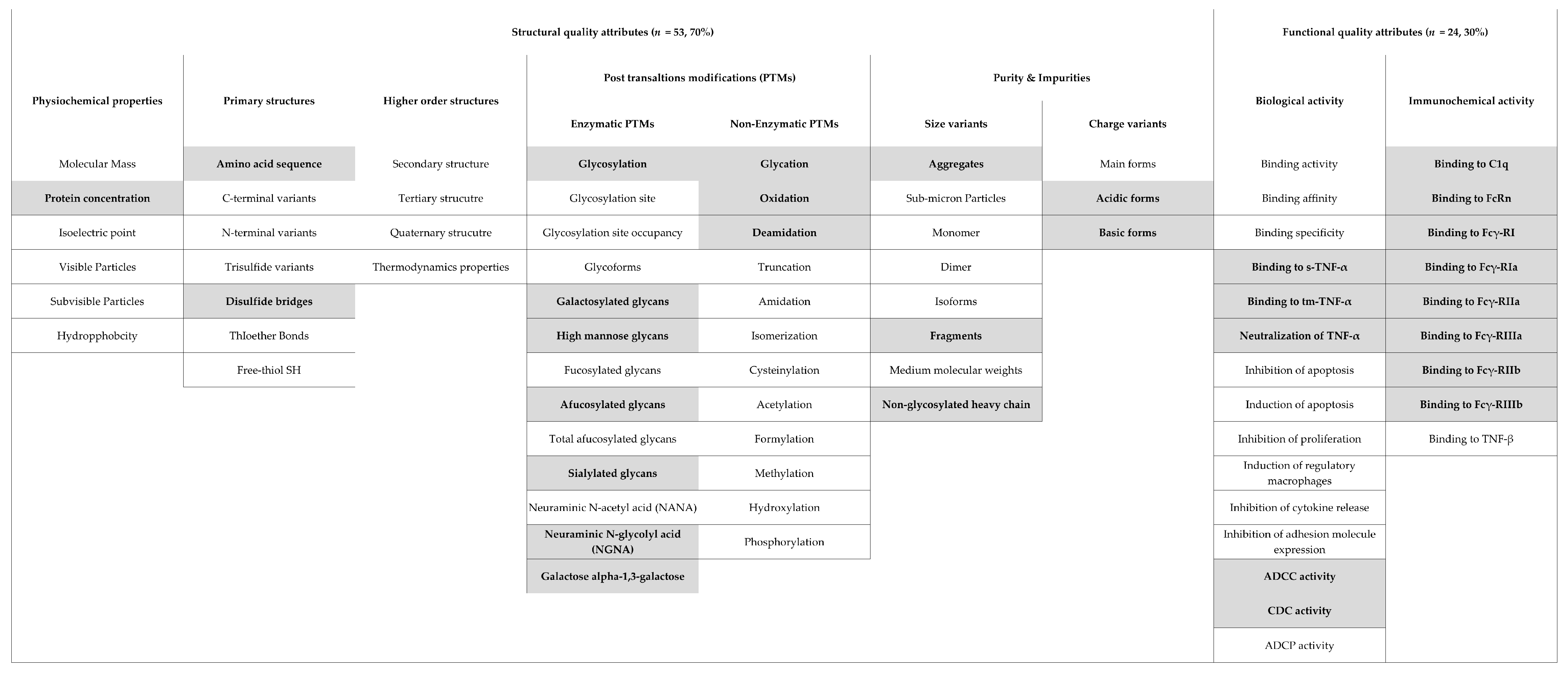
- Regulators often observed minor differences in structural attributes, most commonly in glycoforms and charge variants, between the biosimilar and reference biological, though this had no effect on the functions and clinical profiles and did not preclude biosimilarity.
- Regulators provided a biosimilarity interpretation but rarely reported test results for QAs in EPARs, impeding the interpretation by EPAR users.
1. Introduction
2. Results
2.1. Characteristics of the Included European Public Assessment Reports of Adalimumab Biosimilars

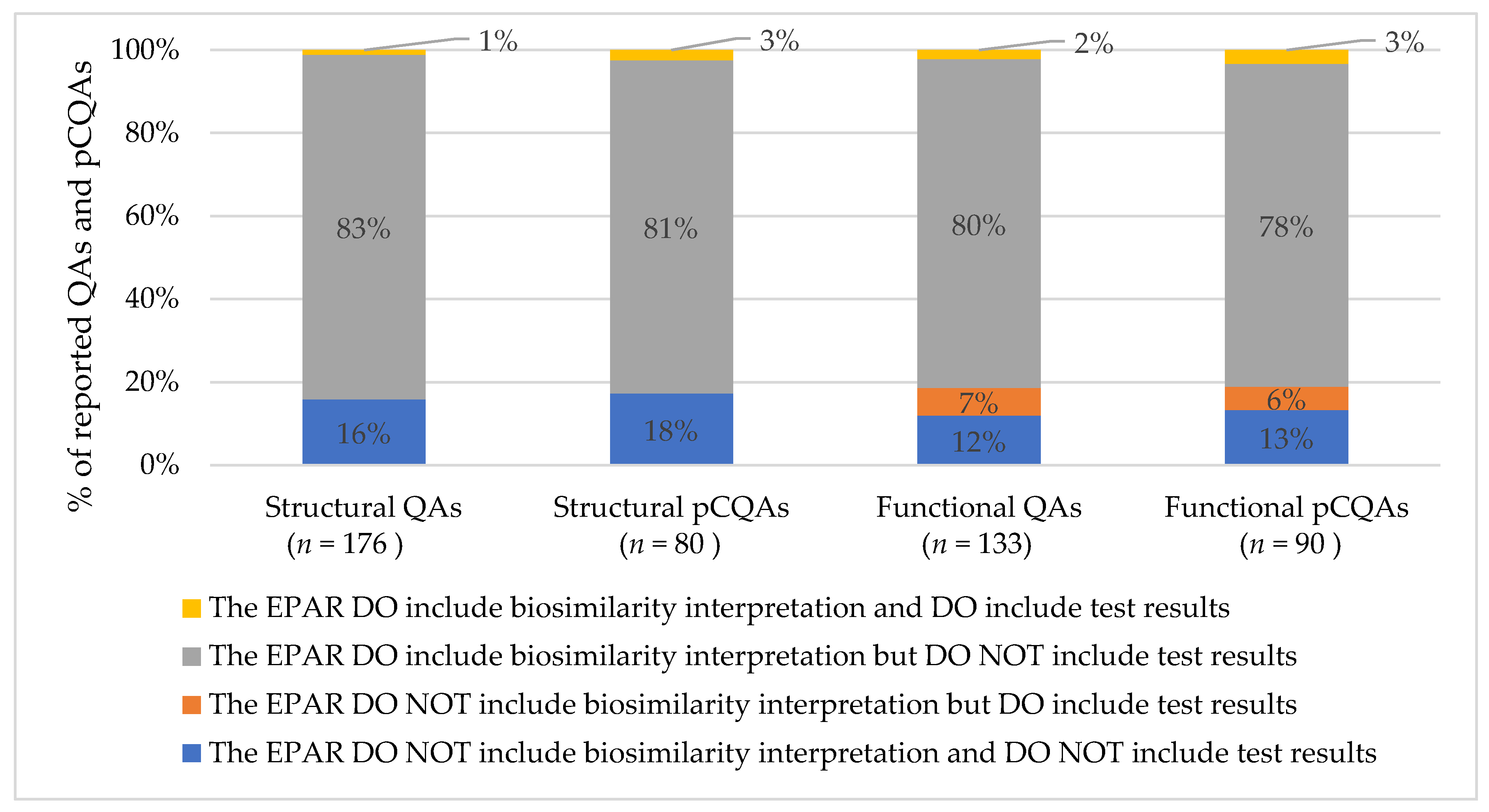{kind=link}
{kind=link}
| Company Code | Date of Initial EPAR Publication (mm/yyyy) | Brand Names | EU Member State of Rapporteurs (Rapporteur and Co-Rapporteur) |
|---|---|---|---|
| ABP501 | 04-2017 | Amgevita® Solymbic® * | Sweden and Italy |
| SB5 | 08-2017 | Imraldi® | Finland and Austria |
| BI695501 | 11-2017 | Cyltezo® * | Austria and Germany |
| GP2017 | 08-2018 | Hefiya® Halimatoz® Hyrimoz® | Austria and Ireland |
| FKB327 | 09-2018 | Hulio® | Belgium and United Kingdom |
| MSB11022 | 04-2019 | Idacio® Kromeya® * | Netherlands and Lithuania |
| PF06410293 | 02-2020 | Amsparity® | Finland and Romania |
2.2. Types of Reported (Potentially Critical) Quality Attributes
2.3. Extent of Information on Reported (Potentially Critical) Quality Attributes
3. Discussion
4. Methods
4.1. Study Cohort
4.2. Information on (Potentially Critical) Quality Attributes in EPARs
4.2.1. Types of Reported (Potentially Critical) Quality Attributes
4.2.2. Extent of Reported Information on (Potentially Critical) Quality Attributes
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Schiestl, M.; Stangler, T.; Torella, C.; Cepeljnik, T.; Toll, H.; Grau, R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat. Biotechnol. 2011, 29, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Planinc, A.; Dejaegher, B.; Vander Heyden, Y.; Viaene, J.; Van Praet, S.; Rappez, F.; Van Antwerpen, P.; Delporte, C. Batch-to-batch N-glycosylation study of infliximab, trastuzumab and bevacizumab, and stability study of bevacizumab. Eur. J. Hosp. Pharm. 2017, 24, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Moorkens, E.; Vulto, A.G.; Huys, I. Biosimilars: Regulatory Frameworks for Marketing Authorization of Biosimilars: Where Do We Go from Here. EPLR 2018, 2, 31. [Google Scholar] [CrossRef]
- EMA. Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues; European Medicines Agency (EMA)-Publications: Amsterdam, The Netherlands, 2014. [Google Scholar]
- EMA. Guideline on Similar Biological Medicinal Products; European Medicines Agency (EMA)-Publications: Amsterdam, The Netherlands, 2015. [Google Scholar]
- USFDA. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations; US Food and Drug Adminstration (USFDA)-Publications: Silver Spring, MD, USA, 2019.
- WHO. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs); World Health Orgnization (WHO)-Publications: Geneva, Switzerland, 2009. [Google Scholar]
- Schiestl, M.; Ranganna, G.; Watson, K.; Jung, B.; Roth, K.; Capsius, B.; Trieb, M.; Bias, P.; Marechal-Jamil, J. The Path Towards a Tailored Clinical Biosimilar Development. BioDrugs 2020, 34, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.J.; Wong, A.C.; Woollett, G.R. An Efficient Development Paradigm for Biosimilars. BioDrugs 2019, 33, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Wolff-Holz, E.; Tiitso, K.; Vleminckx, C.; Weise, M. Evolution of the EU Biosimilar Framework: Past and Future. BioDrugs 2019, 33, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Bielsky, M.C.; Cook, A.; Wallington, A.; Exley, A.; Kauser, S.; Hay, J.L.; Both, L.; Brown, D. Streamlined approval of biosimilars: Moving on from the confirmatory efficacy trial. Drug Discov. Today 2020, 25, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- EMA. ICH Guideline Q8 (R2) on Pharmaceutical Development Step 5; International Conference on Harmonization (ICH)-Publications: Geneva, Switzerland, 2015. [Google Scholar]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252. [Google Scholar] [CrossRef]
- Kim, S.; Song, J.; Park, S.; Ham, S.; Paek, K.; Kang, M.; Chae, Y.; Seo, H.; Kim, H.C.; Flores, M. Drifts in ADCC-related quality attributes of Herceptin®: Impact on development of a trastuzumab biosimilar. MAbs 2017, 9, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Pivot, X.; Pegram, M.; Cortes, J.; Lüftner, D.; Lyman, G.H.; Curigliano, G.; Bondarenko, I.; Yoon, Y.C.; Kim, Y.; Kim, C. Three-year follow-up from a phase 3 study of SB3 (a trastuzumab biosimilar) versus reference trastuzumab in the neoadjuvant setting for human epidermal growth factor receptor 2-positive breast cancer. Eur. J. Cancer 2019, 120, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Vulto, A.G.; Jaquez, O.A. The process defines the product: What really matters in biosimilar design and production? Rheumatology 2017, 56, iv14–iv29. [Google Scholar] [CrossRef] [Green Version]
- Eon-Duval, A.; Broly, H.; Gleixner, R. Quality attributes of recombinant therapeutic proteins: An assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol. Prog 2012, 28, 608–622. [Google Scholar] [CrossRef]
- Kwon, O.; Joung, J.; Park, Y.; Kim, C.W.; Hong, S.H. Considerations of critical quality attributes in the analytical comparability assessment of biosimilar products. Biologicals 2017, 48, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, K.; Seidl, A.; Gutka, H.; Kumar, M.; Gratzl, G.; Keire, D.; Coffey, T.; Kuehne, H. Rational Selection, Criticality Assessment, and Tiering of Quality Attributes and Test Methods for Analytical Similarity Evaluation of Biosimilars. AAPS J. 2018, 20, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, J.; Seamon, K.; Venugopal, S. A-MAb: A Case Study in Bioprocess Development; CASSS and ISPE, CMC Biotech Working Group: Emeryville, CA, USA, 2009; pp. 1–278. [Google Scholar]
- Vessely, C.; Bussineau, C. QbD in biopharmaceutical manufacturing and biosimilar development. In Biosimilars; Springer: Berlin/Heidelberg, Germany, 2018; pp. 187–219. [Google Scholar]
- Tebbey, P.W.; Varga, A.; Naill, M.; Clewell, J.; Venema, J. Consistency of quality attributes for the glycosylated monoclonal antibody Humira® (adalimumab). MAbs 2015, 7, 805–811. [Google Scholar] [CrossRef]
- Liu, J.; Eris, T.; Li, C.; Cao, S.; Kuhns, S. Assessing Analytical Similarity of Proposed Amgen Biosimilar ABP 501 to Adalimumab. BioDrugs 2016, 30, 321–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velayudhan, J.; Chen, Y.F.; Rohrbach, A.; Pastula, C.; Maher, G.; Thomas, H.; Brown, R.; Born, T.L. Demonstration of Functional Similarity of Proposed Biosimilar ABP 501 to Adalimumab. BioDrugs 2016, 30, 339–351. [Google Scholar] [CrossRef]
- Lee, J.J.; Yang, J.; Lee, C.; Moon, Y.; Ahn, S.; Yang, J. Demonstration of functional similarity of a biosimilar adalimumab SB5 to Humira(®). Biologicals 2019, 58, 7–15. [Google Scholar] [CrossRef]
- Lee, N.; Lee, J.J.; Yang, H.; Baek, S.; Kim, S.; Kim, S.; Lee, T.; Song, D.; Park, G. Evaluation of similar quality attribute characteristics in SB5 and reference product of adalimumab. MAbs 2019, 11, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Kronthaler, U.; Fritsch, C.; Hainzl, O.; Seidl, A.; da Silva, A. Comparative functional and pharmacological characterization of Sandoz proposed biosimilar adalimumab (GP2017): Rationale for extrapolation across indications. Expert Opin. Biol. Ther. 2018, 18, 921–930. [Google Scholar] [CrossRef]
- Schreiber, S.; Yamamoto, K.; Muniz, R.; Iwura, T. Physicochemical analysis and biological characterization of FKB327 as a biosimilar to adalimumab. Pharmacol. Res. Perspect. 2020, 8, e00604. [Google Scholar] [CrossRef] [PubMed]
- Magnenat, L.; Palmese, A.; Fremaux, C.; D’Amici, F.; Terlizzese, M.; Rossi, M.; Chevalet, L. Demonstration of physicochemical and functional similarity between the proposed biosimilar adalimumab MSB11022 and Humira®. MAbs 2017, 9, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Derzi, M.; Shoieb, A.M.; Ripp, S.L.; Finch, G.L.; Lorello, L.G.; O’Neil, S.P.; Radi, Z.; Syed, J.; Thompson, M.S.; Leach, M.W. Comparative nonclinical assessments of the biosimilar PF-06410293 and originator adalimumab. Regul. Toxicol. Pharmacol. 2020, 112, 104587. [Google Scholar] [CrossRef]
- Zhang, E.; Xie, L.; Qin, P.; Lu, L.; Xu, Y.; Gao, W.; Wang, L.; Xie, M.H.; Jiang, W.; Liu, S. Quality by Design-Based Assessment for Analytical Similarity of Adalimumab Biosimilar HLX03 to Humira®. AAPS J. 2020, 22, 69. [Google Scholar] [CrossRef]
- Lee, J.F.; Litten, J.B.; Grampp, G. Comparability and biosimilarity: Considerations for the healthcare provider. Curr. Med. Res. Opin. 2012, 28, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.R.; McCombie, R. ICH Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Processes. In ICH Quality Guidelines; Wiley: Hoboken, NJ, USA, 2018; Volume 409. [Google Scholar]
- Ramanan, S.; Grampp, G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs 2014, 28, 363–372. [Google Scholar] [CrossRef]
- van der Plas, R.M.; Hoefnagel, M.H.N.; Hillege, H.L.; Roes, K.C.B. Pragmatic rules for comparability of biological medicinal products. Biologicals 2020, 63, 97–100. [Google Scholar] [CrossRef]
- EMA. Human Medicines; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2020. [Google Scholar]
- AbbVie. HUMIRA (Adalimumab) Injection, Full Prescribing Information; AbbVie Inc.: North Chicago, IL, USA, 2009. [Google Scholar]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Simoens, S.; Vulto, A.G.; Huys, I. European Stakeholder Learnings Regarding Biosimilars: Part II-Improving Biosimilar Use in Clinical Practice. BioDrugs 2020, 34, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Simoens, S.; Vulto, A.G.; Huys, I. European Stakeholder Learnings Regarding Biosimilars: Part I-Improving Biosimilar Understanding and Adoption. BioDrugs 2020, 34, 783–796. [Google Scholar] [CrossRef]
- Moorkens, E.; Jonker-Exler, C.; Huys, I.; Declerck, P.; Simoens, S.; Vulto, A.G. Overcoming Barriers to the Market Access of Biosimilars in the European Union: The Case of Biosimilar Monoclonal Antibodies. Front. Pharmacol. 2016, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, J.; Jilma, B.; Jones, B.; Koenig, F. An update on the clinical evidence that supports biosimilar approvals in Europe. Br. J. Clin. Pharmacol. 2018, 84, 1415–1431. [Google Scholar] [CrossRef]
- Mielke, J.; Jilma, B.; Koenig, F.; Jones, B. Clinical trials for authorized biosimilars in the European Union: A systematic review. Br. J. Clin. Pharmacol. 2016, 82, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Bellinvia, S.; Cummings, J.R.F.; Ardern-Jones, M.R.; Edwards, C.J. Adalimumab Biosimilars in Europe: An Overview of the Clinical Evidence. BioDrugs 2019, 33, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Alsamil, A.M.; Giezen, T.J.; Egberts, T.C.; Leufkens, H.G.; Vulto, A.G.; van der Plas, M.R.; Gardarsdottir, H. Reporting of quality attributes in scientific publications presenting biosimilarity assessments of (intended) biosimilars: A systematic literature review. Eur. J. Pharm. Sci. 2020, 154, 105501. [Google Scholar] [CrossRef] [PubMed]
- Alsamil, A.M.; Giezen, T.J.; Egberts, T.C.; Leufkens, H.G.; Gardarsdottir, H. Comparison of consistency and complementarity of reporting biosimilar quality attributes between regulatory and scientific communities: An adalimumab case study. Biologicals 2021, 69, 30–37. [Google Scholar] [CrossRef]
- EMA. EPAR Amgevita, EMA/CHMP/106922/2017; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2017. [Google Scholar]
- EMA. EPAR Solymbic, EMA/CHMP/106921; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2017. [Google Scholar]
- EMA. EPAR Cyltezo, EMA/CHMP/750187; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2017. [Google Scholar]
- EMA. EPAR Hefiya, EMA/CHMP/520007; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018. [Google Scholar]
- EMA. EPAR Halimatoz, EMA/CHMP/519681; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018. [Google Scholar]
- EMA. EPAR Hyrimoz, EMA/CHMP 404076; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018. [Google Scholar]
- EMA. EPAR Hulio, EMA/CHMP/541826; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2019. [Google Scholar]
- EMA. EPAR Idacio, EMA/CHMP/124342; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2019. [Google Scholar]
- EMA. EPAR Kromeya, EMA/CHMP/214726; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2019. [Google Scholar]
- EMA. EPAR Amsparity, EMA/CHMP/2756; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2020. [Google Scholar]
- EMA. EPAR Imraldi, EMA/CHMP/559383; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2017. [Google Scholar]
- Weise, M.; Kurki, P.; Wolff-Holz, E.; Bielsky, M.C.; Schneider, C.K. Biosimilars: The science of extrapolation. Blood 2014, 124, 3191–3196. [Google Scholar] [CrossRef]
- Ebbers, H.C.; Chamberlain, P. Controversies in Establishing Biosimilarity: Extrapolation of Indications and Global Labeling Practices. BioDrugs 2016, 30, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declerck, P.; Danesi, R.; Petersel, D.; Jacobs, I. The Language of Biosimilars: Clarification, Definitions, and Regulatory Aspects. Drugs 2017, 77, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Tesser, J.R.; Furst, D.E.; Jacobs, I. Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics 2017, 11, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Ebbers, H.C.; Fehrmann, B.; Ottosen, M.; Hvorslev, N.; Høier, P.; Hwang, J.W.; Chung, J.; Lim, H.T.; Lee, S.; Hong, J.; et al. Batch-to-Batch Consistency of SB4 and SB2, Etanercept and Infliximab Biosimilars. BioDrugs 2020, 34, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, P.; Brassart, L.; Blake, P.; Hart, A.; Whitbread, L.; Pembrey, R.; Kieffer, J. Transparency in drug regulation: Public assessment reports in Europe and Australia. Drug Discov. Today 2016, 21, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Cohen, H.; Beydoun, D.; Chien, D.; Lessor, T.; McCabe, D.; Muenzberg, M.; Popovian, R.; Uy, J. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv. Ther. 2016, 33, 2160–2172. [Google Scholar] [CrossRef] [Green Version]
- Hallersten, A.; Fürst, W.; Mezzasalma, R. Physicians prefer greater detail in the biosimilar label (SmPC)–results of a survey across seven European countries. Regul. Toxicol. Pharmacol. 2016, 77, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, L.; Breckenridge, A.; Hoekman, J.; Leufkens, H.; Lumpkin, M.; McAuslane, N.; Stolk, P.; Zhi, K.; Rägo, L. Accelerating access to new medicines: Current status of facilitated regulatory pathways used by emerging regulatory authorities. J. Public Health Policy 2016, 37, 315–333. [Google Scholar] [CrossRef]
- Liberti, L.; Stolk, P.; McAuslane, N.; Somauroo, A.; Breckenridge, A.; Leufkens, H. Adaptive licensing and facilitated regulatory pathways: A survey of stakeholder perceptions. Clin. Pharmacol. Ther. 2015, 98, 477–479. [Google Scholar] [CrossRef]
- Luigetti, R.; Bachmann, P.; Cooke, E.; Salmonson, T. Collaboration, not competition: Developing new reliance models. WHO Drug Inf. 2016, 30, 558. [Google Scholar]
- WHO. Good Regulatory Practices: Guidelines for National Regulatory Authorities for Medical Products; Working Document QAS/16.686. Draft for comment Prepared by EMP/RSS; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Roth, L.; Bempong, D.; Babigumira, J.B.; Banoo, S.; Cooke, E.; Jeffreys, D.; Kasonde, L.; Leufkens, H.G.; Lim, J.C.; Lumpkin, M. Expanding global access to essential medicines: Investment priorities for sustainably strengthening medical product regulatory systems. Glob. Health 2018, 14, 102. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.N.; Thorpe, R.; Knezevic, I.; Casas Levano, M.; Chilufya, M.B.; Chirachanakul, P.; Chua, H.M.; Dalili, D.; Foo, F.; Gao, K.; et al. Regulatory challenges with biosimilars: An update from 20 countries. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef]
- IPRP. The Basics of Analytical Comparability of Biosimilar Monoclonal Antibody for Regulatory Reviewers; The International Pharmaceutical Regulators Programme (IPRP)-Publications: Valparaiso, IN, USA, 2018. [Google Scholar]
- Berntgen, M.; Gourvil, A.; Pavlovic, M.; Goettsch, W.; Eichler, H.-G.; Kristensen, F.B. Improving the contribution of regulatory assessment reports to health technology assessments—A collaboration between the European Medicines Agency and the European network for Health Technology Assessment. Value Health 2014, 17, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Company Code | All QAs (n = 77, 100%) | Type of QAs | All pCQAs (n = 31, 100%) | Type of pCQAs | ||
|---|---|---|---|---|---|---|
| Structural (n = 53, %) | Functional (n = 24, %) | Structural (n = 18, %) | Functional (n = 13, %) | |||
| ABP501 | 36 (47%) | 18 (34%) | 18 (75%) | 20 (65%) | 7 (39%) | 13 (100%) |
| SB5 | 49 (64%) | 27 (51%) | 22 (92%) | 27 (87%) | 14 (78%) | 13 (100%) |
| BI695501 | 27 (35%) | 12 (23%) | 15 (63%) | 20 (65%) | 7 (39%) | 13 (100%) |
| GP2017 | 52 (68%) | 34 (64%) | 18 (75%) | 27 (87%) | 14 (78%) | 13 (100%) |
| FKB327 | 58 (75%) | 39 (74%) | 19 (79%) | 27 (87%) | 14 (78%) | 13 (100%) |
| MSB11022 | 42 (55%) | 20 (38%) | 22 (92%) | 25 (81%) | 12 (67%) | 13 (100%) |
| PF06410293 | 46 (60%) | 27 (51%) | 19 (79%) | 24 (77%) | 12 (67%) | 12 (92%) |
| Consistent for all biosimilars | 16 (21%) | 4 (8%) | 12 (54%) | 16 (52%) | 4 (22%) | 12 (92%) |
| Reporting Catagories | Biosimilarity Interpretation | ||
|---|---|---|---|
| No | Yes | ||
| Test results | No | Reported QAs and pCQAs include no biosimilarity interpretation and no test results, for example:
| Reported QAs and pCQAs include the biosimilarity interpretation but not test results, for example:
|
| Yes | Reported QAs and pCQAs include the test results but not the biosimilarity interpretation, for example:
| Reported QAs and pCQAs include the biosimilarity interpretation and test results, for example,
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsamil, A.M.; Giezen, T.J.; Egberts, T.C.; Leufkens, H.G.; Gardarsdottir, H. Type and Extent of Information on (Potentially Critical) Quality Attributes Described in European Public Assessment Reports for Adalimumab Biosimilars. Pharmaceuticals 2021, 14, 189. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030189
Alsamil AM, Giezen TJ, Egberts TC, Leufkens HG, Gardarsdottir H. Type and Extent of Information on (Potentially Critical) Quality Attributes Described in European Public Assessment Reports for Adalimumab Biosimilars. Pharmaceuticals. 2021; 14(3):189. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030189
Chicago/Turabian StyleAlsamil, Ali M., Thijs J. Giezen, Toine C. Egberts, Hubert G. Leufkens, and Helga Gardarsdottir. 2021. "Type and Extent of Information on (Potentially Critical) Quality Attributes Described in European Public Assessment Reports for Adalimumab Biosimilars" Pharmaceuticals 14, no. 3: 189. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030189