
Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells

 ,
,  ,
,
Abstract
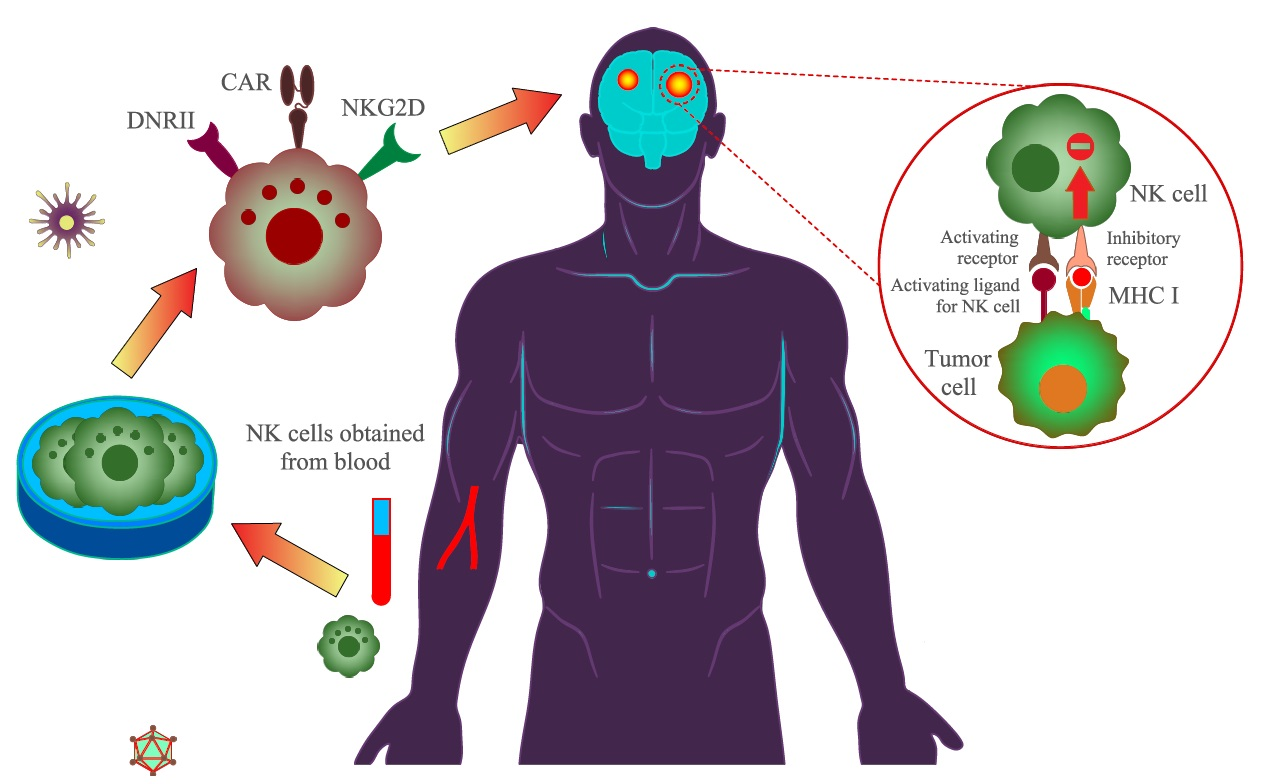
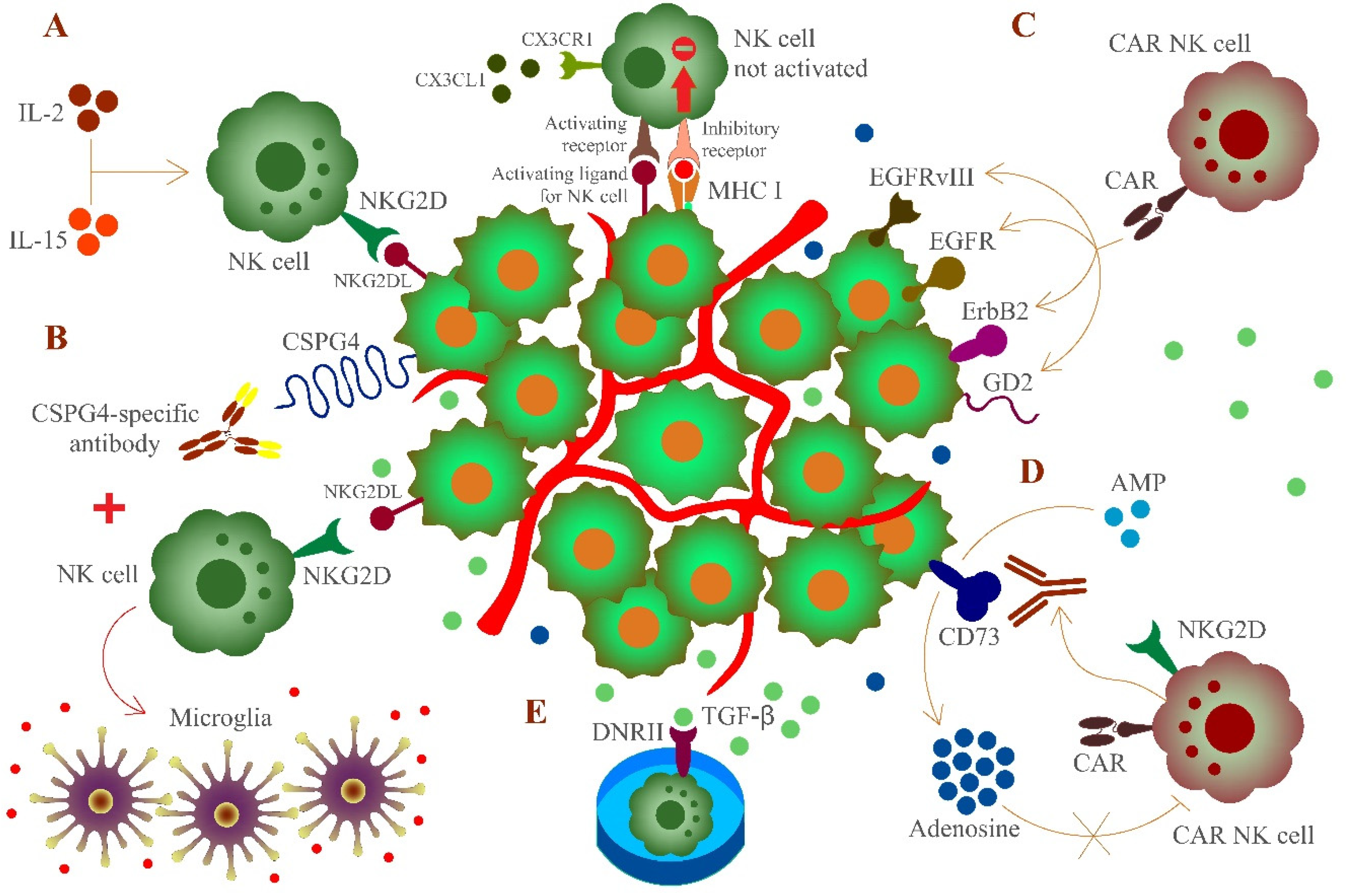
:
{kind=link}
{kind=link}
1. Introduction
2. Defective Antigen Presentation of Tumor Cells Is a Result of Immune Evasion
3. Biology of NK Cell Subsets
3.1. Activating and Inhibitory Receptors of NK Cells
3.2. NK Cell Subsets and Their Function
3.3. Molecular Interaction of the Inhibitory Receptor with MHC I
4. NK Cells as a Possible Immunotherapy Approach to Treat Glioblastoma
5. Proteolytic Regulation of MHC I Molecules
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| APCs | antigen-presenting cells |
| Cat | cathepsin |
| CatG | cathepsin G |
| CCL1 | chemokine C-C motif ligand 1 |
| CSPG4 | chondroitin sulfate proteoglycan 4 |
| DCs | dendritic cells |
| D-MMSN | doxorubicin-loaded magnetic mesoporous silica nanoparticles |
| DNAM-1 | DNAX accessory molecule 1 |
| DNRII | TGF-β-dominant-negative receptor II |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor variant III |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| HLA | human leukocyte antigen |
| IL-2 | interleukin 2 |
| ITAM | immunoreceptor tyrosine-based activation motif |
| KIRs | killer immunoglobulin-like receptors |
| MDSCs | myeloid-derived suppressor cells |
| MHC I | major histocompatibility complex class I |
| NETs | neutrophil extracellular traps |
| MICA | MHC class I chain-related protein A |
| MICB | MHC class I chain-related protein B |
| NCR | natural cytotoxicity receptor |
| NK cells | natural killer cells |
| NKG2D | natural killer group 2D activating receptor |
| NKG2DL | natural killer group 2D ligands |
| NSPs | neutrophil serine proteases |
| PBMCs | peripheral blood mononuclear cells |
| TCR | T cell receptor |
| TGF-β | transforming growth factor beta |
| TGN | trans-Golgi network |
| Th cells | T helper cells |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor α |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Tregs | T regulatory cells |
| ULBP1–6 | UL 16 binding protein 1–6 |
References
- Bischof, J.; Westhoff, M.A.; Wagner, J.E.; Halatsch, M.E.; Trentmann, S.; Knippschild, U.; Wirtz, C.R.; Burster, T. Cancer stem cells: The potential role of autophagy, proteolysis, and cathepsins in glioblastoma stem cells. Tumour Biol. 2017, 39, 1010428317692227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; et al. Considering the Experimental use of Temozolomide in Glioblastoma Research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewett, A.; Kos, J.; Kaur, K.; Safaei, T.; Sutanto, C.; Chen, W.; Wong, P.; Namagerdi, A.K.; Fang, C.; Fong, Y.; et al. Natural Killer Cells: Diverse Functions in Tumor Immunity and Defects in Pre-neoplastic and Neoplastic Stages of Tumorigenesis. Mol. Ther. Oncolytics 2020, 16, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2020. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- McCoach, C.E.; Bivona, T.G. The evolving understanding of immunoediting and the clinical impact of immune escape. J. Thorac. Dis. 2018, 10, 1248–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurewicz, M.M.; Stern, L.J. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics 2019, 71, 171–187. [Google Scholar] [CrossRef]
- Dersh, D.; Holly, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Jelencic, V.; Sestan, M.; Kavazovic, I.; Lenartic, M.; Marinovic, S.; Holmes, T.D.; Prchal-Murphy, M.; Lisnic, B.; Sexl, V.; Bryceson, Y.T.; et al. NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat. Immunol. 2018, 19, 1083–1092. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Baragano Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, 2040. [Google Scholar] [CrossRef] [Green Version]
- Narni-Mancinelli, E.; Vivier, E. Shed NKG2D ligand boosts NK cell immunity. Cell Res. 2015, 25, 651–652. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/beta-catenin signaling pathway. Oncogene 2020, 39, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Shi, F.D.; Ljunggren, H.G.; La Cava, A.; Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 2011, 11, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Wildes, T.J.; Dyson, K.A.; Francis, C.; Wummer, B.; Yang, C.; Yegorov, O.; Shin, D.; Grippin, A.; Dean, B.D.; Abraham, R.; et al. Immune Escape After Adoptive T-cell Therapy for Malignant Gliomas. Clin. Cancer Res. 2020, 26, 5689–5700. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkstrom, N.K.; Ljunggren, H.G.; Michaelsson, J. Emerging insights into natural killer cells in human peripheral tissues. Nat. Rev. Immunol. 2016, 16, 310–320. [Google Scholar] [CrossRef]
- Sondergaard, S.R.; Ullum, H.; Pedersen, B.K. Proliferative and cytotoxic capabilities of CD16+CD56™ and CD16+/™CD56+ natural killer cells. APMIS 2000, 108, 831–837. [Google Scholar] [CrossRef]
- Alter, G.; Suscovich, T.J.; Kleyman, M.; Teigen, N.; Streeck, H.; Zaman, M.T.; Meier, A.; Altfeld, M. Low perforin and elevated SHIP-1 expression is associated with functional anergy of natural killer cells in chronic HIV-1 infection. AIDS 2006, 20, 1549–1551. [Google Scholar] [CrossRef]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.T.; Orange, J.S. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, I.; Sonnerborg, I.; Strunz, B.; Friberg, D.; Cornillet, M.; Hertwig, L.; Ivarsson, M.A.; Bjorkstrom, N.K. 29-Color Flow Cytometry: Unraveling Human Liver NK Cell Repertoire Diversity. Front. Immunol. 2019, 10, 2692. [Google Scholar] [CrossRef]
- Poreba, M.; Groborz, K.M.; Rut, W.; Pore, M.; Snipas, S.J.; Vizovisek, M.; Turk, B.; Kuhn, P.; Drag, M.; Salvesen, G.S. Multiplexed Probing of Proteolytic Enzymes Using Mass Cytometry-Compatible Activity-Based Probes. J. Am. Chem. Soc. 2020, 142, 16704–16715. [Google Scholar] [CrossRef]
- Gärtner, F.; Knippschild, U.; Burster, T. Application of an Activity-Based Probe to Determine Proteolytic Activity of Cell Surface Cathepsin G by Mass Cytometry Data Acquisition. ACS Omega 2020, 5, 28233–28238. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Hudspeth, K.; Silva-Santos, B.; Mavilio, D. Natural cytotoxicity receptors: Broader expression patterns and functions in innate and adaptive immune cells. Front. Immunol. 2013, 4, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaldo, E.; Del Zotto, G.; Della Chiesa, M.; Mingari, M.C.; Moretta, A.; De Maria, A.; Moretta, L. Human NK cell receptors/markers: A tool to analyze NK cell development, subsets and function. Cytom. Part A 2013, 83, 702–713. [Google Scholar] [CrossRef]
- Cichocki, F.; Miller, J.S.; Anderson, S.K.; Bryceson, Y.T. Epigenetic regulation of NK cell differentiation and effector functions. Front. Immunol. 2013, 4, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Verges, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, L.; Pietra, G.; Vacca, P.; Pende, D.; Moretta, F.; Bertaina, A.; Mingari, M.C.; Locatelli, F.; Moretta, A. Human NK cells: From surface receptors to clinical applications. Immunol. Lett. 2016, 178, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.P.; Villa-Alvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez, S. NK Cells in the Treatment of Hematological Malignancies. J. Clin. Med. 2019, 8, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef] [Green Version]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Moretta, L. Dissecting CD56dim human NK cells. Blood 2010, 116, 3689–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.A.; Laugier-Anfossi, F.; Vely, F.; Saulquin, X.; Riedmuller, J.; Tisserant, A.; Gauthier, L.; Romagne, F.; Ferracci, G.; Arosa, F.A.; et al. Recognition of peptide-MHC class I complexes by activating killer immunoglobulin-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 13224–13229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Marsh, S.G.; Parham, P.; Dupont, B.; Geraghty, D.E.; Trowsdale, J.; Middleton, D.; Vilches, C.; Carrington, M.; Witt, C.; Guethlein, L.A.; et al. Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Immunogenetics 2003, 55, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.G.; Sun, P.D. The structural basis of ligand recognition by natural killer cell receptors. J. Biomed. Biotechnol. 2011, 2011, 203628. [Google Scholar] [CrossRef] [Green Version]
- Chewning, J.H.; Gudme, C.N.; Hsu, K.C.; Selvakumar, A.; Dupont, B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J. Immunol. 2007, 179, 854–868. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, O.; Zimmer, J.; Enger, P.O.; Chekenya, M. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 2014, 193, 6192–6206. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Freud, A.G.; Caligiuri, M.A. Human natural killer cell development. Immunol. Rev. 2006, 214, 56–72. [Google Scholar] [CrossRef]
- Sivori, S.; Cantoni, C.; Parolini, S.; Marcenaro, E.; Conte, R.; Moretta, L.; Moretta, A. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur. J. Immunol. 2003, 33, 3439–3447. [Google Scholar] [CrossRef]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16− killer Ig-like receptor− NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Clements, C.S.; Lin, J.; Sullivan, L.C.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H.; et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, B.K.; Barahmand-Pour, F.; Paulsene, W.; Medley, S.; Geraghty, D.E.; Strong, R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005, 174, 2878–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyington, J.C.; Motyka, S.A.; Schuck, P.; Brooks, A.G.; Sun, P.D. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 2000, 405, 537–543. [Google Scholar] [CrossRef]
- Fan, Q.R.; Long, E.O.; Wiley, D.C. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol. 2001, 2, 452–460. [Google Scholar] [CrossRef]
- Hilton, H.G.; Guethlein, L.A.; Goyos, A.; Nemat-Gorgani, N.; Bushnell, D.A.; Norman, P.J.; Parham, P. Polymorphic HLA-C Receptors Balance the Functional Characteristics of KIR Haplotypes. J. Immunol. 2015, 195, 3160–3170. [Google Scholar] [CrossRef] [Green Version]
- Hilton, H.G.; Vago, L.; Older Aguilar, A.M.; Moesta, A.K.; Graef, T.; Abi-Rached, L.; Norman, P.J.; Guethlein, L.A.; Fleischhauer, K.; Parham, P. Mutation at positively selected positions in the binding site for HLA-C shows that KIR2DL1 is a more refined but less adaptable NK cell receptor than KIR2DL3. J. Immunol. 2012, 189, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Vivian, J.P.; Duncan, R.C.; Berry, R.; O’Connor, G.M.; Reid, H.H.; Beddoe, T.; Gras, S.; Saunders, P.M.; Olshina, M.A.; Widjaja, J.M.; et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 2011, 479, 401–405. [Google Scholar] [CrossRef]
- Orr, M.T.; Murphy, W.J.; Lanier, L.L. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat. Immunol. 2010, 11, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.M.; Mahmoud, A.B.; Makrigiannis, A.P. Licensed and Unlicensed NK Cells: Differential Roles in Cancer and Viral Control. Front. Immunol. 2016, 7, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertwig, L.; Hamann, I.; Romero-Suarez, S.; Millward, J.M.; Pietrek, R.; Chanvillard, C.; Stuis, H.; Pollok, K.; Ransohoff, R.M.; Cardona, A.E.; et al. CX3CR1-dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur. J. Immunol. 2016, 46, 1984–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Siminska, D.; Kojder, K.; Grochans, S.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Fractalkine/CX3CL1 in Neoplastic Processes. Int. J. Mol. Sci. 2020, 21, 3723. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M.; Simon, P.; Loffler, C.; Capper, D.; Bunz, B.; Harter, P.; Schlaszus, H.; Schleich, A.; Tabatabai, G.; Goeppert, B.; et al. Elevated HLA-E levels in human glioblastomas but not in grade I to III astrocytomas correlate with infiltrating CD8+ cells. J. Neuroimmunol. 2007, 189, 50–58. [Google Scholar] [CrossRef]
- Wischhusen, J.; Friese, M.A.; Mittelbronn, M.; Meyermann, R.; Weller, M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: Implications for immune escape in vivo. J. Neuropathol. Exp. Neurol. 2005, 64, 523–528. [Google Scholar] [CrossRef]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Domingues, P.; Gonzalez-Tablas, M.; Otero, A.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Goncalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.H.; Teodosio, C.; Ortiz, J.; Sousa, P.; Otero, A.; Maillo, A.; Barcena, P.; Garcia-Macias, M.C.; Lopes, M.C.; de Oliveira, C.; et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am. J. Pathol. 2012, 181, 1749–1761. [Google Scholar] [CrossRef]
- Friese, M.A.; Platten, M.; Lutz, S.Z.; Naumann, U.; Aulwurm, S.; Bischof, F.; Buhring, H.J.; Dichgans, J.; Rammensee, H.G.; Steinle, A.; et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003, 63, 8996–9006. [Google Scholar] [PubMed]
- Wu, A.; Wiesner, S.; Xiao, J.; Ericson, K.; Chen, W.; Hall, W.A.; Low, W.C.; Ohlfest, J.R. Expression of MHC I and NK ligands on human CD133+ glioma cells: Possible targets of immunotherapy. J. Neurooncol. 2007, 83, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Los, M.J.; Azizi, Z.; et al. HSP70/IL-2 Treated NK Cells Effectively Cross the Blood Brain Barrier and Target Tumor Cells in a Rat Model of Induced Glioblastoma Multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluschke, G.; Vanek, M.; Evans, A.; Dittmar, T.; Schmid, P.; Itin, P.; Filardo, E.J.; Reisfeld, R.A. Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc. Natl. Acad. Sci. USA 1996, 93, 9710–9715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol. Biol. Lett. 2017, 22, 4. [Google Scholar] [CrossRef] [Green Version]
- Pellegatta, S.; Savoldo, B.; Di Ianni, N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, A.; Wang, J.; Domingues, O.; Planaguma, J.; Yan, T.; Rygh, C.B.; Skaftnesmo, K.O.; Thorsen, F.; McCormack, E.; Hentges, F.; et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget 2013, 4, 1527–1546. [Google Scholar] [CrossRef] [Green Version]
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology 2013, 2, e23401. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel Human NK Cell Line Carrying CAR Targeting EGFRvIII Induces Antitumor Effects in Glioblastoma Cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longee, D.C.; Wikstrand, C.J.; Mansson, J.E.; He, X.; Fuller, G.N.; Bigner, S.H.; Fredman, P.; Svennerholm, L.; Bigner, D.D. Disialoganglioside GD2 in human neuroectodermal tumor cell lines and gliomas. Acta Neuropathol. 1991, 82, 45–54. [Google Scholar] [CrossRef]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Wang, J.; Matosevic, S. NT5E/CD73 as Correlative Factor of Patient Survival and Natural Killer Cell Infiltration in Glioblastoma. J. Clin. Med. 2019, 8, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J. Hematol. Oncol. 2020, 13, 76. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Waller-Wise, R. Umbilical cord blood: Information for childbirth educators. J. Perinat. Educ. 2011, 20, 54–60. [Google Scholar] [CrossRef]
- Dalle, J.H.; Menezes, J.; Wagner, E.; Blagdon, M.; Champagne, J.; Champagne, M.A.; Duval, M. Characterization of cord blood natural killer cells: Implications for transplantation and neonatal infections. Pediatr. Res. 2005, 57, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Phan, M.T.; Lee, S.H.; Kim, S.K.; Cho, D. Expansion of NK Cells Using Genetically Engineered K562 Feeder Cells. Methods Mol. Biol. 2016, 1441, 167–174. [Google Scholar] [CrossRef]
- Vasu, S.; Berg, M.; Davidson-Moncada, J.; Tian, X.; Cullis, H.; Childs, R.W. A novel method to expand large numbers of CD56(+) natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy 2015, 17, 1582–1593. [Google Scholar] [CrossRef]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Montealegre, S.; van Endert, P.M. Endocytic Recycling of MHC Class I Molecules in Non-professional Antigen Presenting and Dendritic Cells. Front. Immunol. 2018, 9, 3098. [Google Scholar] [CrossRef]
- Machold, R.P.; Ploegh, H.L. Intermediates in the assembly and degradation of class I major histocompatibility complex (MHC) molecules probed with free heavy chain-specific monoclonal antibodies. J. Exp. Med. 1996, 184, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.G.; Hengel, H.; Koszinowski, U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palesch, D.; Wagner, J.; Meid, A.; Molenda, N.; Sienczyk, M.; Burkhardt, J.; Munch, J.; Prokop, L.; Stevanovic, S.; Westhoff, M.A.; et al. Cathepsin G-mediated proteolytic degradation of MHC class I molecules to facilitate immune detection of human glioblastoma cells. Cancer Immunol. Immunother. 2016, 65, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Craik, C.S.; Page, M.J.; Madison, E.L. Proteases as therapeutics. Biochem. J. 2011, 435, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Schernberg, A.; Nivet, A.; Dhermain, F.; Ammari, S.; Escande, A.; Pallud, J.; Louvel, G.; Deutsch, E. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin. Transl. Radiat. Oncol. 2018, 10, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.J.; Nannuru, K.C.; Futakuchi, M.; Sadanandam, A.; Singh, R.K. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res. 2008, 68, 5803–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, E.J.; Lo, J.H.; Bhatia, S.N. Smart nanosystems: Bio-inspired technologies that interact with the host environment. Proc. Natl. Acad. Sci. USA 2015, 112, 14460–14466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alphandery, E. Nano-Therapies for Glioblastoma Treatment. Cancers 2020, 12, 242. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, H.; Tie, C.; Yan, C.; Deng, Z.; Wan, Q.; Liu, X.; Yan, F.; Zheng, H. MR imaging tracking of inflammation-activatable engineered neutrophils for targeted therapy of surgically treated glioma. Nat. Commun. 2018, 9, 4777. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Hempel, A.; Janiszewski, T.; Kolt, S.; Snipas, S.J.; Drag, M.; Salvesen, G.S. NETosis Occurs Independently of Neutrophil Serine Proteases. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burster, T.; Gärtner, F.; Bulach, C.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030236
Burster T, Gärtner F, Bulach C, Zhanapiya A, Gihring A, Knippschild U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals. 2021; 14(3):236. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030236
Chicago/Turabian StyleBurster, Timo, Fabian Gärtner, Christiane Bulach, Anuar Zhanapiya, Adrian Gihring, and Uwe Knippschild. 2021. "Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells" Pharmaceuticals 14, no. 3: 236. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030236