
Internalization of Foldamer-Based DNA Mimics through a Site-Specific Antibody Conjugate to Target HER2-Positive Cancer Cells
 , , , , and
, , , , and
Abstract
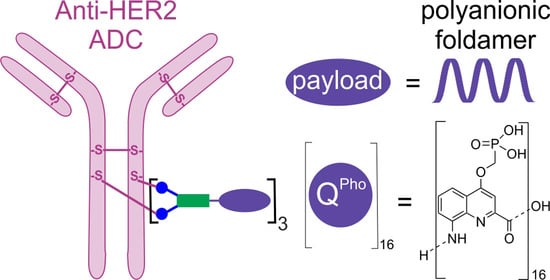
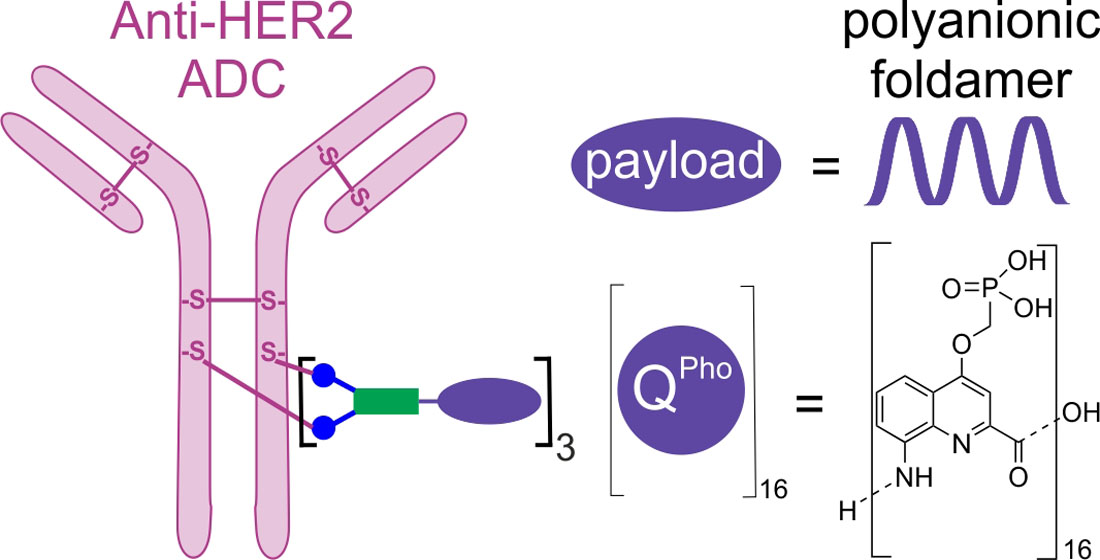
:
1. Introduction
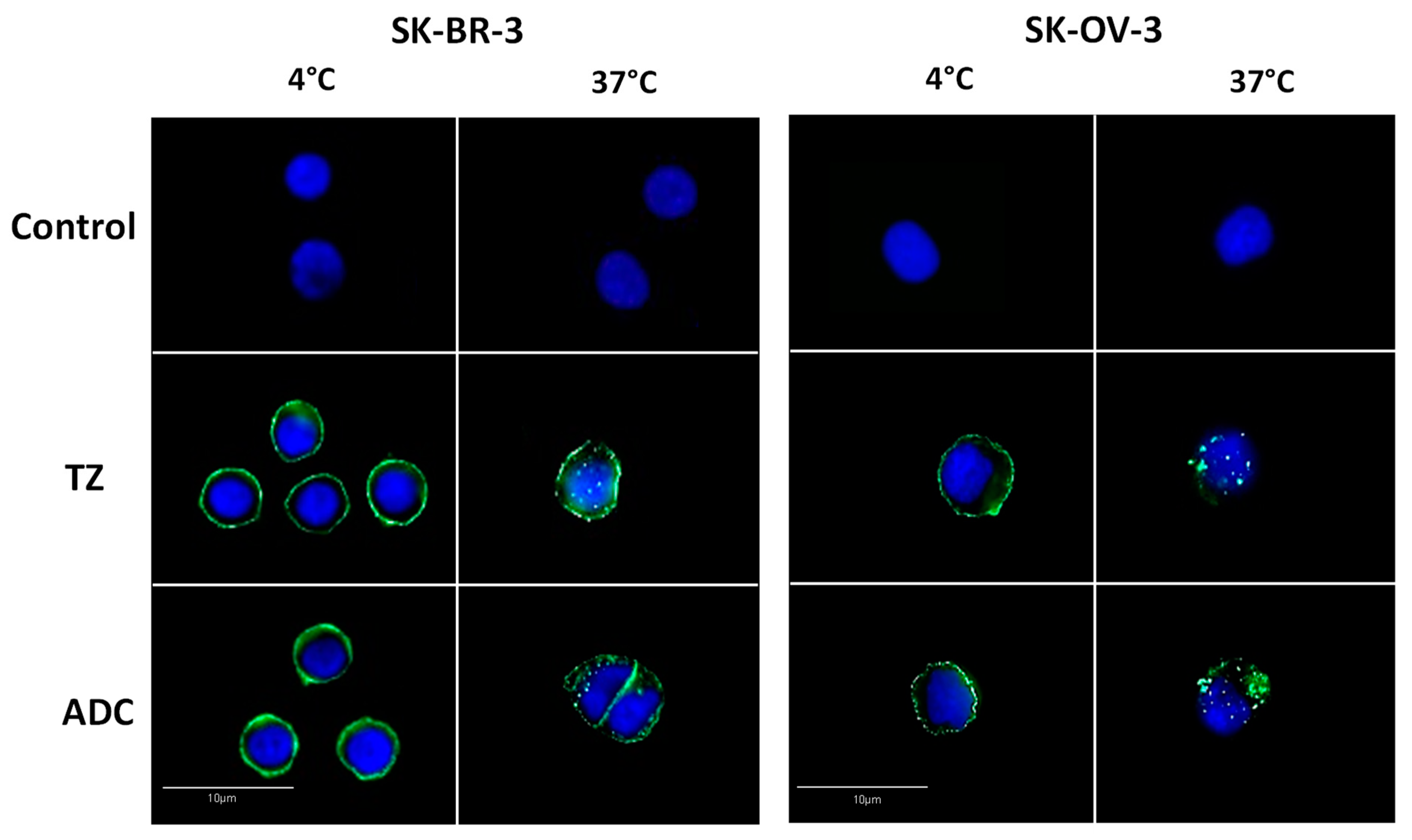
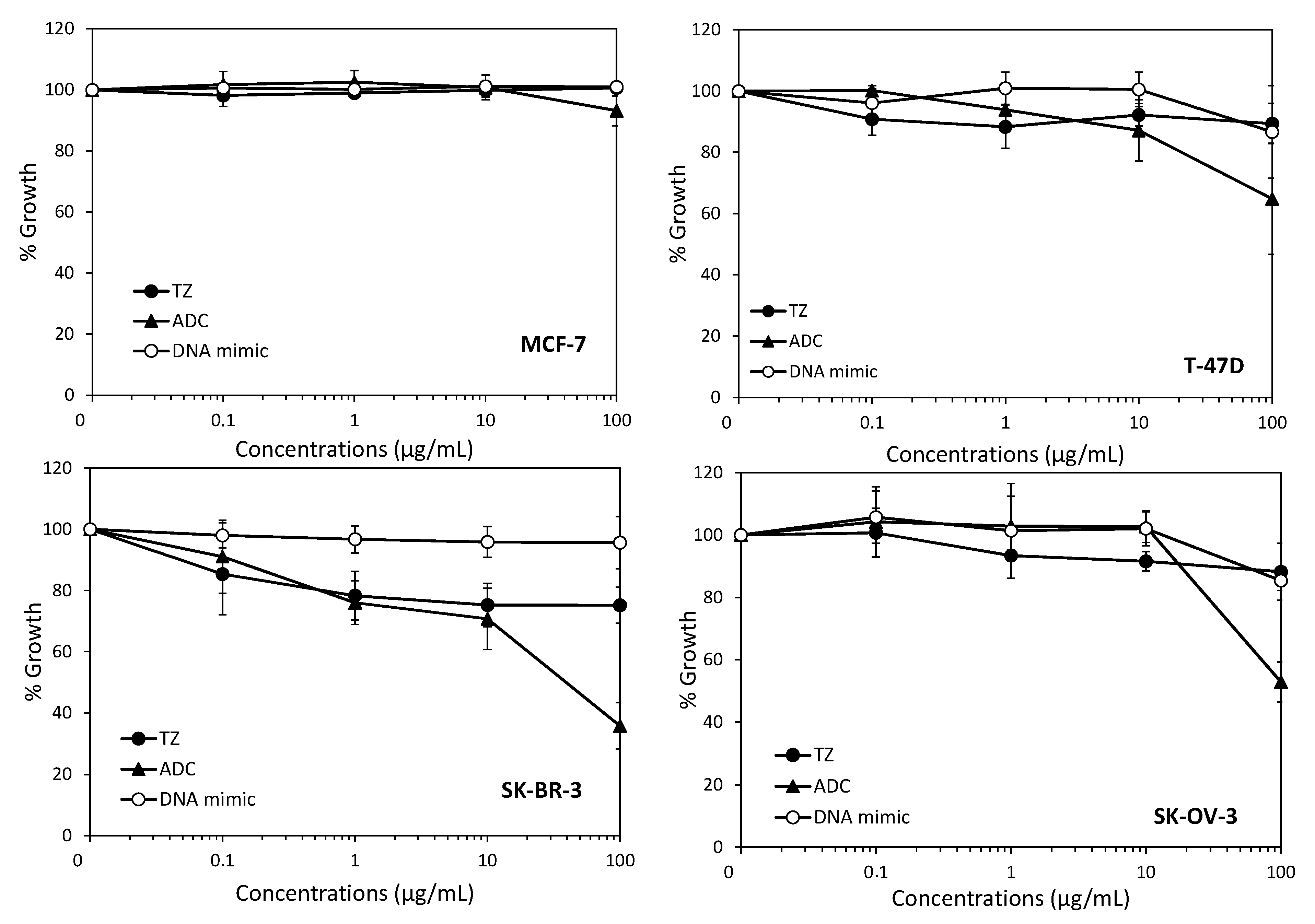
2. Results and Discussion
3. Materials and Methods
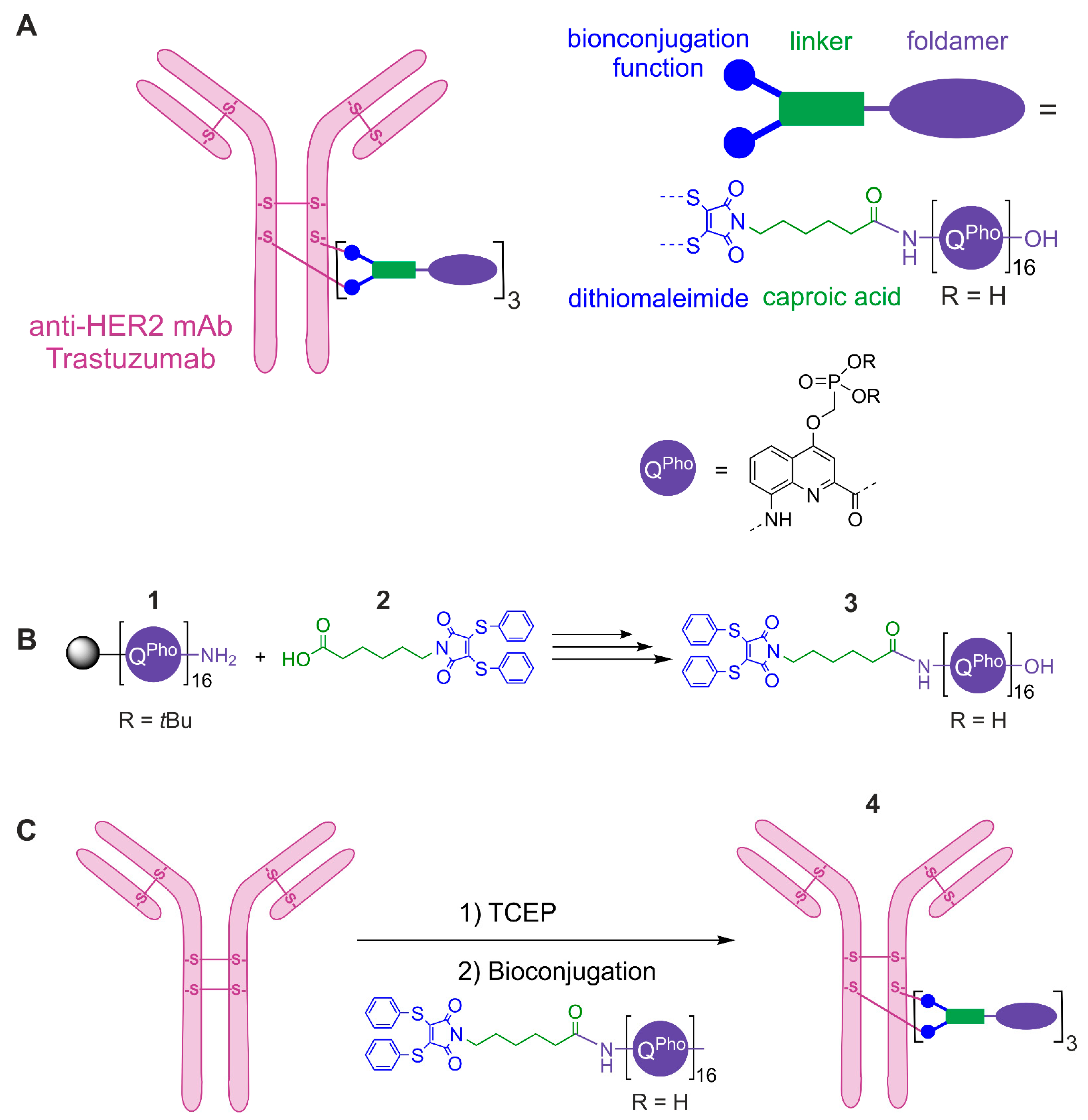
3.1. Chemical Synthesis and Characterization of a Modified DNA Mimic Foldamer
3.2. Preparation of Quinoline Monomer and Solid Phase Synthesis of Oligomers 1 and 3
3.3. Bioconjugation, Purification and Characterization of the ADC 4
3.4. Cell Culture
3.5. Cell Growth Inhibition Assay
3.6. Analysis of Antibody-Drug Conjugate Internalization by Immunofluorescence
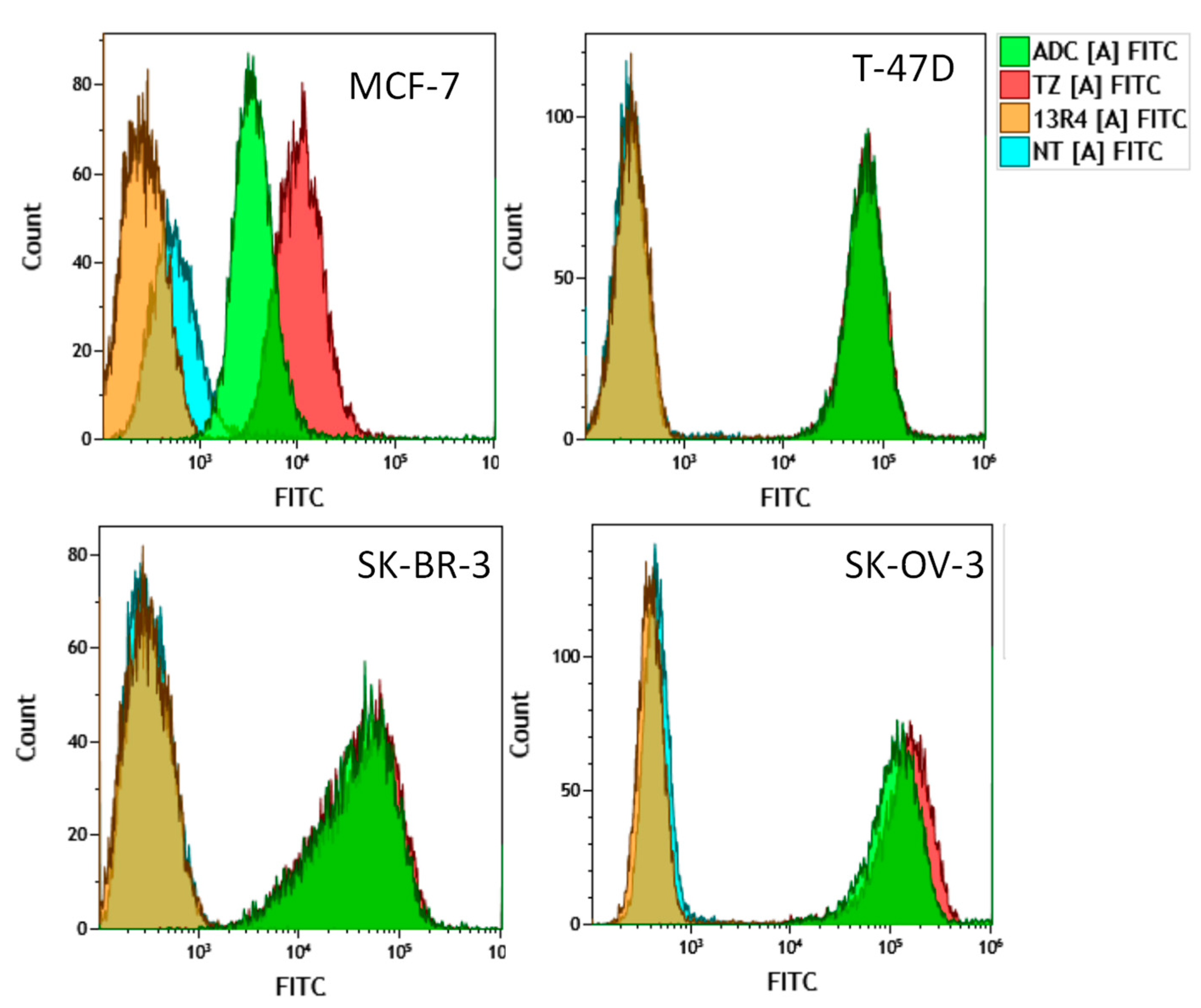
3.7. Flow Cytometry Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, C.; Awasthi, S.K. Versatility of peptide nucleic acids (PNAs): Role in chemical biology, drug discovery and origins of life. Chem. Biol. Drug Des. 2017, 89, 16–37. [Google Scholar] [CrossRef]
- Papargyri, N.; Pontoppidan, M.; Andersen, M.R.; Koch, T.; Hagedorn, P.H. Chemical Diversity of Locked Nucleic Acid-Modified Antisense Oligonucleotides Allows Optimization of Pharmaceutical Properties. Mol. Ther. Nucleic Acids 2020, 19, 706–717. [Google Scholar] [CrossRef]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acids: Promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010, 7, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Kawamoto, Y.; Hashiya, F.; Hashiya, K.; Yamamoto, M.; Kizaki, S.; Bando, T.; Sugiyama, H. Sequence-specific DNA alkylation and transcriptional inhibition by long-chain hairpin pyrrole-imidazole polyamide-chlorambucil conjugates targeting CAG/CTG trinucleotide repeats. Bioorg. Med. Chem. 2014, 22, 4646–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.S.; Meier, J.L.; Dervan, P.B. Design of sequence-specific DNA binding molecules for DNA methyltransferase inhibition. J. Am. Chem. Soc. 2014, 136, 3687–3694. [Google Scholar] [CrossRef]
- Zhang, Y.; Sicot, G.; Cui, X.; Vogel, M.; Wuertzer, C.A.; Lezon-Geyda, K.; Wheeler, J.; Harki, D.A.; Muzikar, K.A.; Stolper, D.A.; et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole-imidazole polyamide. Biochemistry 2011, 50, 10431–10441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Marchand, C. Interfacial inhibitors of protein-nucleic acid interactions. Curr. Med. Chem. Anti Cancer Agents 2005, 5, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Morishita, R.; Gibbons, G.H.; Horiuchi, M.; Ellison, K.E.; Nakama, M.; Zhang, L.; Kaneda, Y.; Ogihara, T.; Dzau, V.J. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation In Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 5855–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, M.; Prokoph, N.; Girbig, M.; Wang, X.; Huang, Y.-H.; Srivastava, Y.; Hou, L.; Narasimhan, K.; Kolatkar, P.R.; Francois, M.; et al. Structure and decoy-mediated inhibition of the SOX18/Prox1-DNA interaction. Nucleic Acids Res. 2016, 44, 3922–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, M.; Wagner, A.H. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017, 144, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, D.; Bianco, P.R.; Kumar, K. De novo design of protein mimics of B-DNA. Mol. Biosyst. 2016, 12, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-C.; Ho, C.-H.; Hsu, K.-C.; Yang, J.-M.; Wang, A.H.-J. DNA mimic proteins: Functions, structures, and bioinformatic analysis. Biochemistry 2014, 53, 2865–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziach, K.; Chollet, C.; Parissi, V.; Prabhakaran, P.; Marchivie, M.; Corvaglia, V.; Bose, P.P.; Laxmi-Reddy, K.; Godde, F.; Schmitter, J.-M.; et al. Single helically folded aromatic oligoamides that mimic the charge surface of double-stranded B-DNA. Nat. Chem. 2018, 10, 511–518. [Google Scholar] [CrossRef]
- Corvaglia, V.; Carbajo, D.; Prabhakaran, P.; Ziach, K.; Mandal, P.K.; Santos, V.D.; Legeay, C.; Vogel, R.; Parissi, V.; Pourquier, P.; et al. Carboxylate-functionalized foldamer inhibitors of HIV-1 integrase and Topoisomerase 1: Artificial analogues of DNA mimic proteins. Nucleic Acids Res. 2019, 47, 5511–5521. [Google Scholar] [CrossRef] [Green Version]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharm. Basel. Switz. 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet Lond. Engl. 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Dokter, W.; Ubink, R.; van der Lee, M.; van der Vleuten, M.; van Achterberg, T.; Jacobs, D.; Loosveld, E.; van den Dobbelsteen, D.; Egging, D.; Mattaar, E.; et al. Preclinical profile of the HER2-targeting ADC SYD983/SYD985: Introduction of a new duocarmycin-based linker-drug platform. Mol. Cancer Ther. 2014, 13, 2618–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, N.; Denevault-Sabourin, C.; Bryden, F.; Viaud-Massuard, M.-C. Towards antibody-drug conjugates and prodrug strategies with extracellular stimuli-responsive drug delivery in the tumor microenvironment for cancer therapy. Eur. J. Med. Chem. 2017, 142, 393–415. [Google Scholar] [CrossRef]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C.; et al. Impact of cathepsin B-sensitive triggers and hydrophilic linkers on in vitro efficacy of novel site-specific antibody-drug conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, N.; Allard-Vannier, E.; Martin, C.; Bryden, F.; Letast, S.; Colas, C.; Lakhrif, Z.; Collinet, N.; Dimier-Poisson, I.; Chourpa, I.; et al. Site-Specific Conjugation of Auristatins onto Engineered scFv Using Second Generation Maleimide to Target HER2-positive Breast Cancer in Vitro. Bioconjug. Chem. 2018, 29, 3516–3521. [Google Scholar] [CrossRef]
- Nunes, J.P.M.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.B.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody-drug conjugate (ADC). Chem. Commun. Camb. Engl. 2015, 51, 10624–10627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Ghosez, L.; Haveaux, B.; Viehe, H.G. Alkyl and Aryl α-Chloro Enamines. Angew. Chem. Int. Ed. 1969, 8, 454–455. [Google Scholar] [CrossRef]
- Dawson, S.J.; Hu, X.; Claerhout, S.; Huc, I. Solid Phase Synthesis of Helically Folded Aromatic Oligoamides. Methods Enzymol. 2016, 580, 279–301. [Google Scholar]
- Hu, X.; Dawson, S.J.; Mandal, P.K.; de Hatten, X.; Baptiste, B.; Huc, I. Optimizing side chains for crystal growth from water: A case study of aromatic amide foldamers. Chem. Sci. 2017, 8, 3741–3749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptiste, B.; Douat-Casassus, C.; Laxmi-Reddy, K.; Godde, F.; Huc, I. Solid phase synthesis of aromatic oligoamides: Application to helical water-soluble foldamers. J. Org. Chem. 2010, 75, 7175–7185. [Google Scholar] [CrossRef]
- Hu, X.; Dawson, S.J.; Nagaoka, Y.; Tanatani, A.; Huc, I. Solid-Phase Synthesis of Water-Soluble Helically Folded Hybrid α-Amino Acid/Quinoline Oligoamides. J. Org. Chem. 2016, 81, 1137–1150. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release Off. J. Control. Release Soc. 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Bäumer, N.; Berdel, W.E.; Bäumer, S. Immunoprotein-Mediated siRNA Delivery. Mol. Pharm. 2017, 14, 1339–1351. [Google Scholar] [CrossRef]
- Jung, S.; Ohk, J.; Jeong, D.; Li, C.; Lee, S.; Duan, J.; Kim, C.; Lim, J.-S.; Yang, Y.; Kim, K.-I.; et al. Distinct regulatory effect of the p34SEI-1 oncoprotein on cancer metastasis in HER2/neu-positive and -negative cells. Int. J. Oncol. 2014, 45, 189–196. [Google Scholar] [CrossRef]
- Ludyga, N.; Englert, S.; Pflieger, K.; Rauser, S.; Braselmann, H.; Walch, A.; Auer, G.; Höfler, H.; Aubele, M. The impact of cysteine-rich intestinal protein 1 (CRIP1) in human breast cancer. Mol. Cancer 2013, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Spänkuch, B.; Heim, S.; Kurunci-Csacsko, E.; Lindenau, C.; Yuan, J.; Kaufmann, M.; Strebhardt, K. Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer Res. 2006, 66, 5836–5846. [Google Scholar] [CrossRef] [Green Version]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, C.D.; De Mazière, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.; Li, G.; Dugger, D.L.; Crocke, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Audette, C.; Hoffee, M.; Lambert, J.M.; Blättler, W.A. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J. Pharmacol Exp. Ther. 2004, 308, 1073–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwkowski, M.X.; Lofgren, J.A.; Lewis, G.D.; Hotaling, T.E.; Fendly, B.M.; Fox, J.A. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin. Oncol. 1999, 26, 60–70. [Google Scholar]
- Wang, H.; Wang, W.; Xu, Y.; Yang, Y.; Chen, X.; Quan, H.; Lou, L. Aberrant intracellular metabolism of T-DM1 confers T-DM1 resistance in human epidermal growth factor receptor 2-positive gastric cancer cells. Cancer Sci. 2017, 108, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Cini, E.; Faltoni, V.; Petricci, E.; Taddei, M.; Salvini, L.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Anastasi, A.M.; Battistuzzi, G.; et al. Antibody drug conjugates (ADCs) charged with HDAC inhibitor for targeted epigenetic modulation. Chem. Sci. 2018, 9, 6490–6496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milazzo, F.M.; Vesci, L.; Anastasi, A.M.; Chiapparino, C.; Rosi, A.; Giannini, G.; Taddei, M.; Cini, E.; Faltoni, V.; Petricci, E.; et al. ErbB2 Targeted Epigenetic Modulation: Anti-tumor Efficacy of the ADC Trastuzumab-HDACi ST8176AA1. Front. Oncol. 2019, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13R4 | TZ | ADC | |
|---|---|---|---|
| MCF-7 | 0.45 (0.6, 0.3) | 13.1 (14.1, 12.2) | 6.7 (7.2, 6.2) |
| T-47D | 0.4 (0.3, 0.5) | 85.0 (65.9, 104.0) | 83.6 (65.1, 102.0) |
| SK-BR-3 | 0.4 (0.3, 0.45) | 88.0 (89.0, 87.0) | 84.6 (86.1, 83.0) |
| SK-OV-3 | 0.5 (0.5, 0.5) | 223.5 (222.0, 225.0) | 180.0 (177.0, 183.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corvaglia, V.; Ait Mohamed Amar, I.; Garambois, V.; Letast, S.; Garcin, A.; Gongora, C.; Del Rio, M.; Denevault-Sabourin, C.; Joubert, N.; Huc, I.; et al. Internalization of Foldamer-Based DNA Mimics through a Site-Specific Antibody Conjugate to Target HER2-Positive Cancer Cells. Pharmaceuticals 2021, 14, 624. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070624
Corvaglia V, Ait Mohamed Amar I, Garambois V, Letast S, Garcin A, Gongora C, Del Rio M, Denevault-Sabourin C, Joubert N, Huc I, et al. Internalization of Foldamer-Based DNA Mimics through a Site-Specific Antibody Conjugate to Target HER2-Positive Cancer Cells. Pharmaceuticals. 2021; 14(7):624. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070624
Chicago/Turabian StyleCorvaglia, Valentina, Imène Ait Mohamed Amar, Véronique Garambois, Stéphanie Letast, Aurélie Garcin, Céline Gongora, Maguy Del Rio, Caroline Denevault-Sabourin, Nicolas Joubert, Ivan Huc, and et al. 2021. "Internalization of Foldamer-Based DNA Mimics through a Site-Specific Antibody Conjugate to Target HER2-Positive Cancer Cells" Pharmaceuticals 14, no. 7: 624. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070624