
Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors

1
The First Affiliated Hospital and College of Clinical Medicine of Henan University of Science and Technology, Luoyang 471003, China
2
College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
3
School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
4
School of Nursing, Henan University of Science and Technology, 263 Kaiyuan Road, Luoyang 471003, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Pharmaceuticals 2022, 15(11), 1316; https://0-doi-org.brum.beds.ac.uk/10.3390/ph15111316
Submission received: 10 October 2022
/
Revised: 19 October 2022
/
Accepted: 20 October 2022
/
Published: 25 October 2022
(This article belongs to the Special Issue Click Reactions in Medicinal Chemistry)
Abstract
:Indoleamine 2,3-dioxygenase 1 (IDO1) has received much attention as an immunomodulatory enzyme in the field of cancer immunotherapy. While several IDO1 inhibitors have entered clinical trials, there are currently no IDO1 inhibitor drugs on the market. To explore potential IDO1 inhibitors, we designed a series of compounds with urea and 1,2,3-triazole structures. Organic synthesis and IDO1 enzymatic activity experiments verified the molecular-level activities of the designed compounds, and the IC50 value of compound 3a was 0.75 μM. Molecular docking and quantum mechanical studies further explained the binding mode and reaction potential of compound 3a with IDO1. Our research has resulted in a series of novel IDO1 inhibitors, which is beneficial to the development of drugs targeting IDO1 in numerous cancer diseases.
1. Introduction
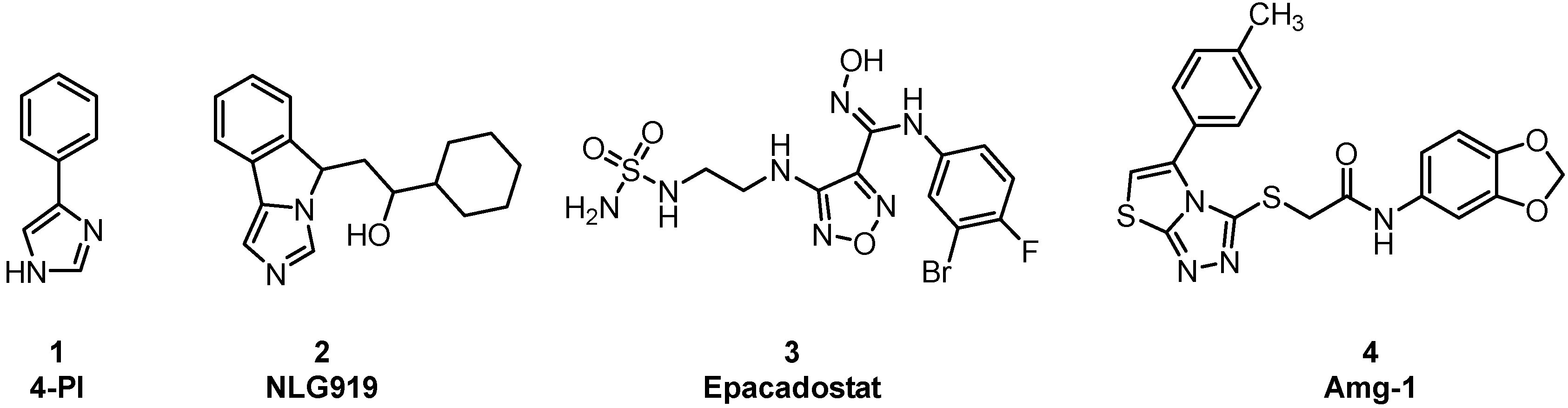
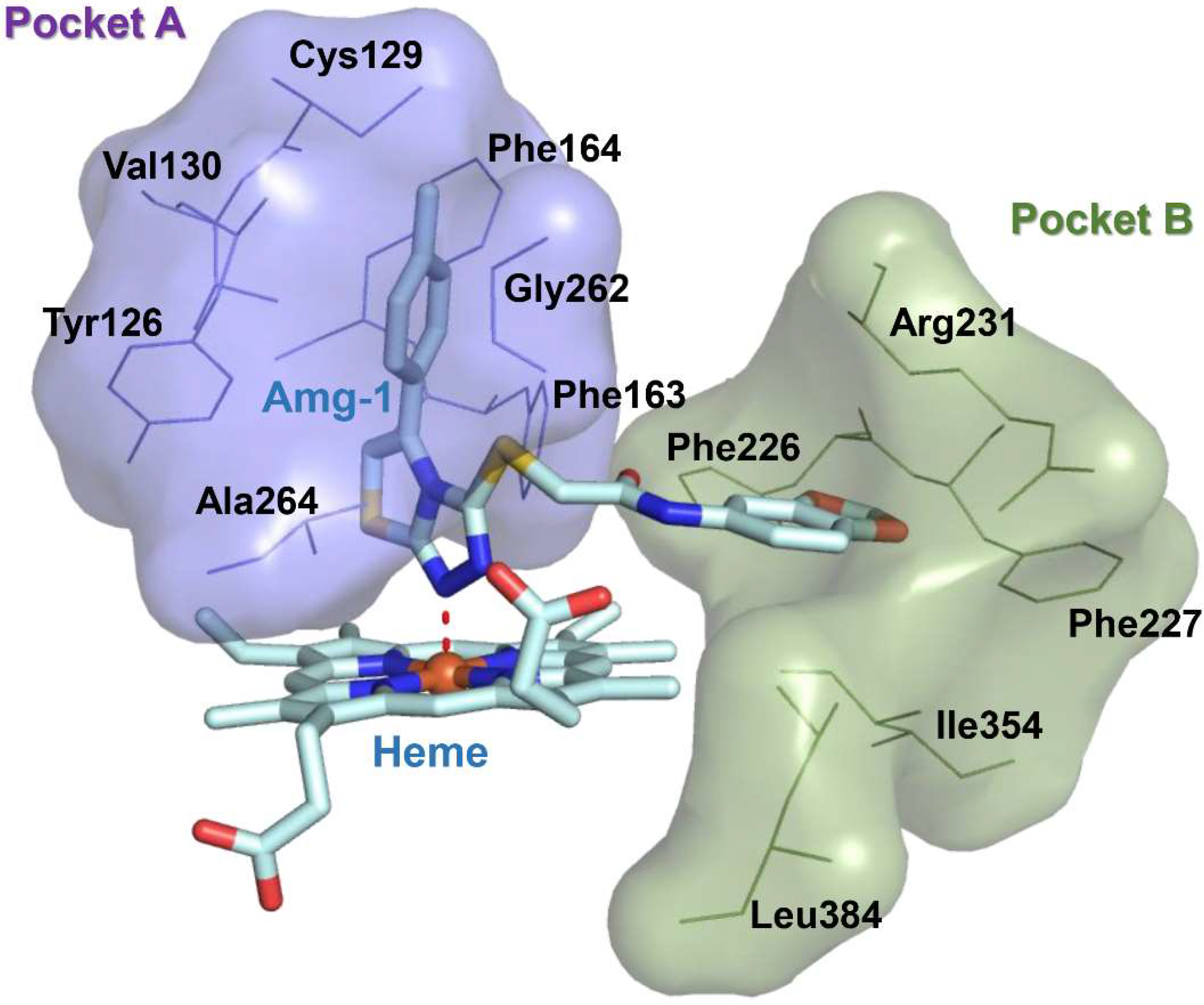
Immuno-oncology is an important form of tumor treatment, along with surgery, radiotherapy, and chemotherapy [1]. It works by activating and mobilizing the body’s own immune system through positive regulation of the immune system, subsequently achieving the capture and clearance of tumor cells [2]. At present, immuno-oncology is largely performed in clinical situations via immune checkpoint inhibition, tumor vaccine therapy, and adoptive cell therapy. Immune checkpoint inhibitors (PD1/PD-L1) have become first-line treatments for advanced non–small-cell lung cancer and melanoma [3]. As another important immune checkpoint, IDO1 overexpression, is observable in many types of tumor cells, such as those of gastric cancer, breast cancer, and brain cancer. IDO1 is a monomeric protein enzyme containing heme [4]. Its main biological function is to catalyze the oxidative cleavage of the L-tryptophan (L-Trp) indole ring to produce kynurenine (KYN). The consumption of L-Trp blocks the proliferation of T-cells and promotes the differentiation of T-cells into regulatory T-cells [5]. KYN metabolites can combine with aryl hydrocarbon receptors (AhRs) to activate signaling pathways, enhancing immune tolerance and leading to immune escape. The toxicity of accumulated metabolites also causes T-cell apoptosis. Consequently, IDO1 inhibitors have attracted the attention of scientists as potential cancer therapeutic drugs. Currently, a few drugs have entered the clinical trial stage, such as 4-PI (Figure 1,1), NLG919 (Figure 1,2), Epacadostat (Figure 1,3), and Amg-1 (Figure 1,4) [6]. Amg-1 is a selective IDO1 inhibitor with better performance than TDO2 and IDO2. The IC50 of Amg-1 was found to be 3.0 μM by the Bridge-It tryptophan fluorescence assay. Our group further analyzed the crystal structure of an Amg-1/IDO1 complex (PDB: 4PK5) and uncovered additional characteristics affecting enzyme recognition by small molecules [7]. Pockets A and B are formed by ligand-induced conformational rearrangement of the residue binding sites located in the catalytic cleft (Figure 2). Of these, pocket A is largely composed of heme and the amino acid residues Tyr126, Cys129, Val130, Phe163, Phe164, Gly262, and Ala264, while pocket B is located at the entrance of the catalytic site and is composed of the residues Phe226, Phe227, Arg231, Ile354, and Leu384. IDO1 inhibitors specifically occupy either pocket A alone or both pockets [8]. This observation confirms the presence of conformational changes in the catalytic site induced by multiple ligands and supports the molecular recognition of enzymes toward inhibitors with different structures. Aryl, halogen-substituted aryl, or heteroaromatic groups (such as indole) are most often chosen to occupy pocket A, while pocket B contains a mixture of hydrophilic (Arg231) and hydrophobic (Phe226) residues [9]. Hence, it is important in the development of small-molecule inhibitors to understand the structural composition of both catalytic pockets of the IDO1 enzymes and their interaction with IDO1 inhibitors.
Urea compounds are used as enzyme inhibitors and lead compounds to develop biomimetic peptides because of their peptide bonds (CONH group) and biological activities akin to those of peptides [10]. These characteristics render them important chemicals in pesticides and medicine, such as herbicides [11], insecticides, bactericides [12,13], and plant-growth regulation [14]. The ability of peptide bonds in urea compounds to combine with enzymes through hydrogen bonding could be manipulated to synthesize cyclin-dependent kinases to block the cell cycle [15]. At present, urea compounds are largely obtained by the direct reaction of isocyanate compounds and amine compounds [16]. Our group performed condensation reactions on differentially substituted phenyl isocyanates and alkyne-terminated amines and obtained, through click chemistry, compounds with urea and 1,2,3-triazole structures.
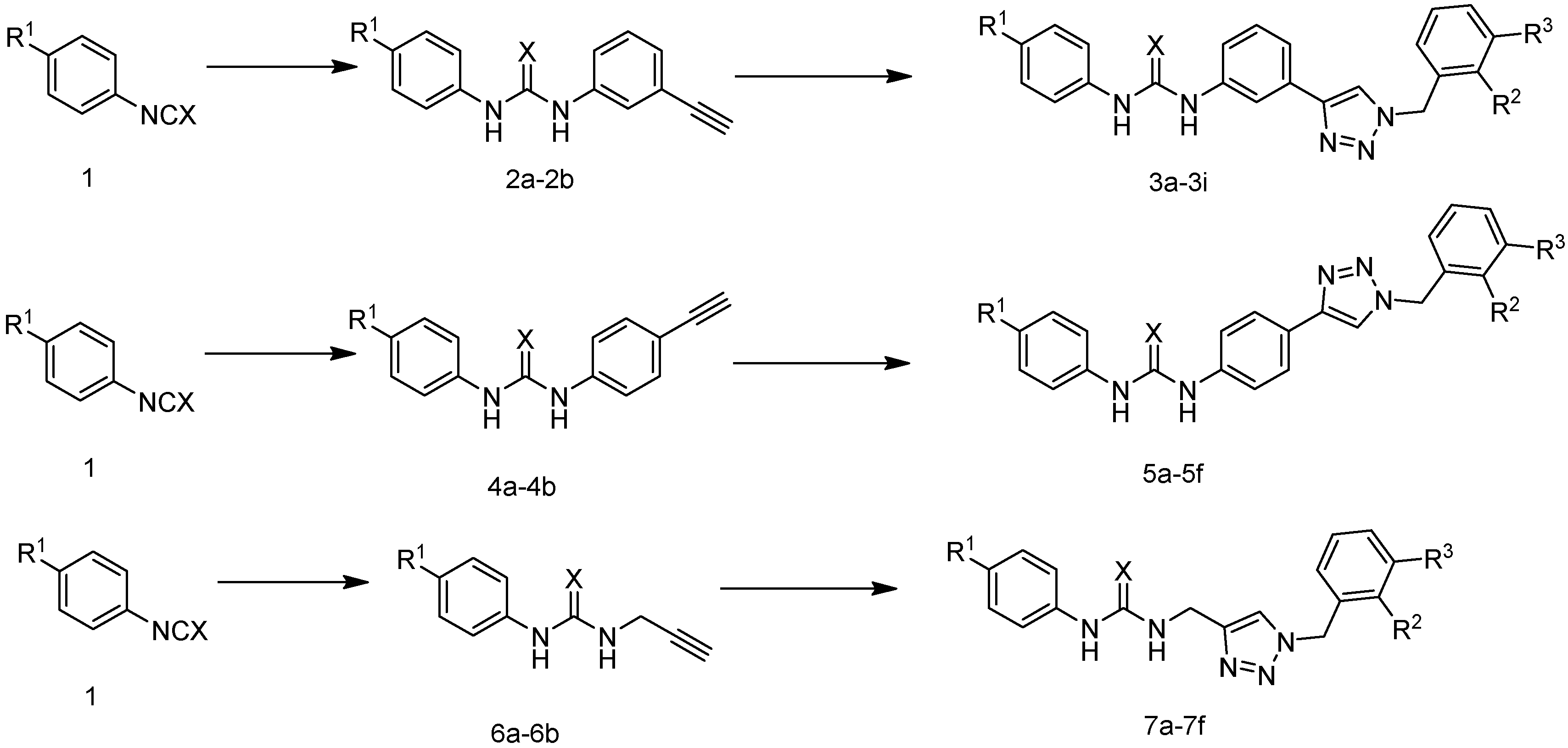
Through comprehensive literature research, our group designed and synthesized a series of novel compounds modeled after the binding mechanism between Amg-1 and the IDO1 enzyme, replacing the oxadiazole structure with the 1,2,3-triazole structure and the hydroxylamine structure with a urea structure. We selected three types of functional groups: m-phenyl group, p-phenyl group, and methyl group as Linkers to connect urea and 1,2,3-triazole blocks respectively. The reaction routes of the three series of compounds (3a–3i, 5a–5f, 7a–7f) are shown in Figure 3 and Table 1. The action conditions of the step were convenient and easy to control. Different substituents (R1, R2, and R3) were present on the benzene ring of phenyl isocyanate and benzyl structures. The effect of linker substituents and R1, R2, and R3 on IDO1 inhibition were compared.
2. Results and Discussion
2.1. IDO1 Inhibition Study
Through literature surveys, we employed the Hela cell-based functional assay to study the IDO1 inhibition activities of the designed compounds. The IC50 value of BMS-986205 as a positive control was 0.62 nM (data not shown), which is consistent with the results previously reported in the literature (IC50 = 0.5 nM) [2]. As shown in Table 2, the activities of compounds 3a–3i against IDO1 were all below 10 μM, of which 3a reached 0.75 μM. The activity of most of compounds 5a–5f and 7a–7f were higher than 10 μM, indicating that the activity of m-phenyl as the linking substituent of urea and triazole was better than that of p-phenyl and methyl. We further compared the effects of different linking groups on the activity by selecting compounds with the same substituents on the left and right benzene rings. Then three groups of compounds (i.e., 3a, 5e and 7f; 3e, 5d and 7e; and 3g, 5c and 7d) were compared to verify that m-phenyl is the better linker substituent in terms of promoting inhibitor activity. It was also found by comparing 3f, 5b and 7b that the three compounds with the same benzene ring substituents on their phenyl isocyanate and benzyl structures but different linker substituents have similar inhibitory activities. This indicates that benzene ring substituents R1 = OCH3, R2 = H, and R3 = Br, all give synthesized compounds good inhibitory activities.
2.2. Molecular Docking Studies of Compounds 3a, 5e and 7f
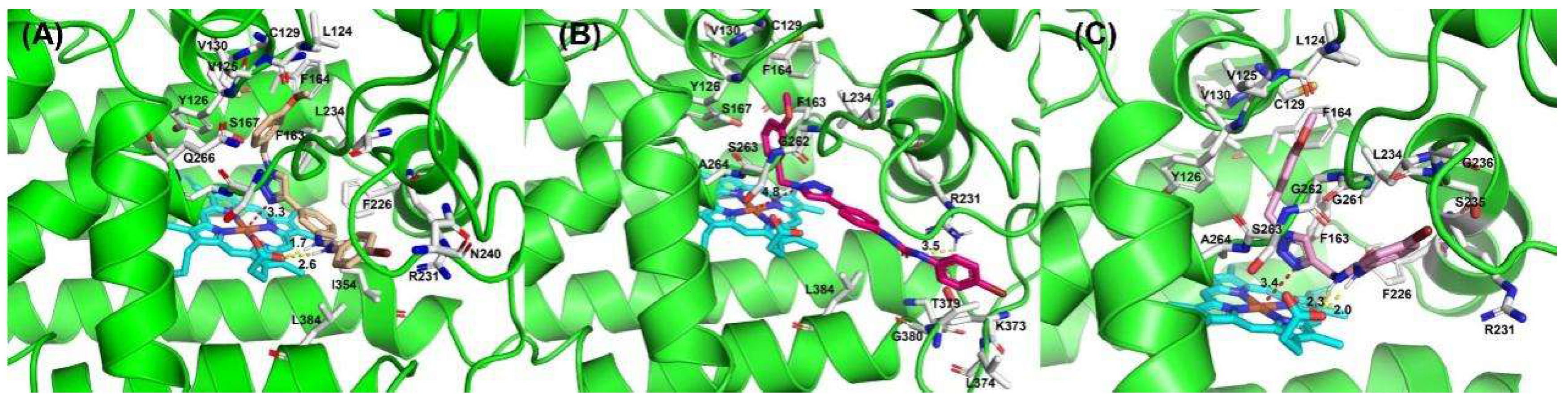
In order to intuitively analyze the binding mode of active compounds to IDO1, we selected compounds 3a, 5e and 7f with different orders of magnitude activity as model compounds. As shown in Figure 4, the docking scores of compounds 3a, 5e and 7f with IDO1 were −7.812, −6.216 and −7.815, respectively. The 1,2,3-triazolyl group in the structures of compounds 3a and 7f was located above the heme and the distance from the ferrous ion in the middle of the heme is 3.33 Å and 3.43 Å, respectively. The binding modes of the compounds are similar to several reported IDO1 inhibitors. The 1,2,3-triazole group in the structure of compound 5e has a distance of 4.82 Å from the heme ferrous ion, which is far away from the heme iron and has a weaker affinity for IDO1. The docking results indicated that compounds 3a and 7f were more capable of coordinating with ferrous ions than 5e. The 1,2,3-triazole in the structure of compound 7f is connected to the urea structure through a flexible methylene group, and the steric twist of the methylene group affects the position of compound 7f in the pocket; thus, the inhibitory activity of compound 7f on IDO1 is weaker than that of compound 3a.
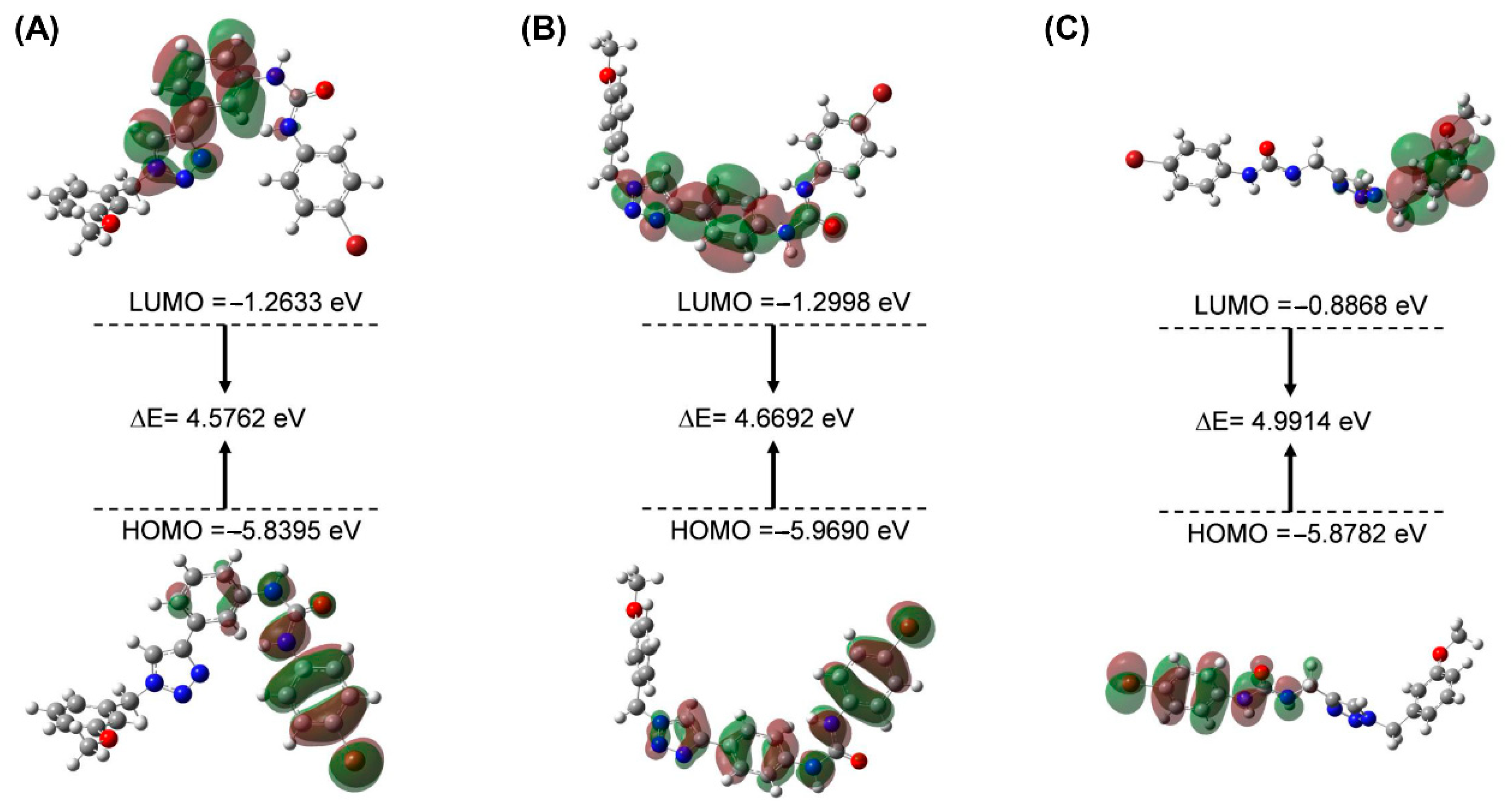
2.3. HOMO-LUMO and MEP Studies
The HOMO and LUMO orbitals of the molecule and the energetic gap values of the orbital characterized the chemical reactions and kinetic stability. The smaller the energetic gap value is, the less stable the compound is, and it is more likely to interact with the protein [17,18]. The energies of Frontier molecular orbitals of compounds 3a, 5e and 7f are shown in Figure 5 and Table 3. The energy gap values of the three compounds of HOMO-LUMO were, respectively, 4.5762, 4.6692, and 4.9914 eV, indicating that the chemical reactions of the compound were 3a > 5e > 7f. The result showed that compound 3a is more likely to interact with the IDO1 protein, followed by 5e; 7f is the worst. Moreover, the calculated results were in good agreement with the experimental results. Furthermore, the urea group in compounds 3a and 5e was located in the HOMO orbitals, which provided electrons for the amino acid interaction at the IDO1 binding pocket.
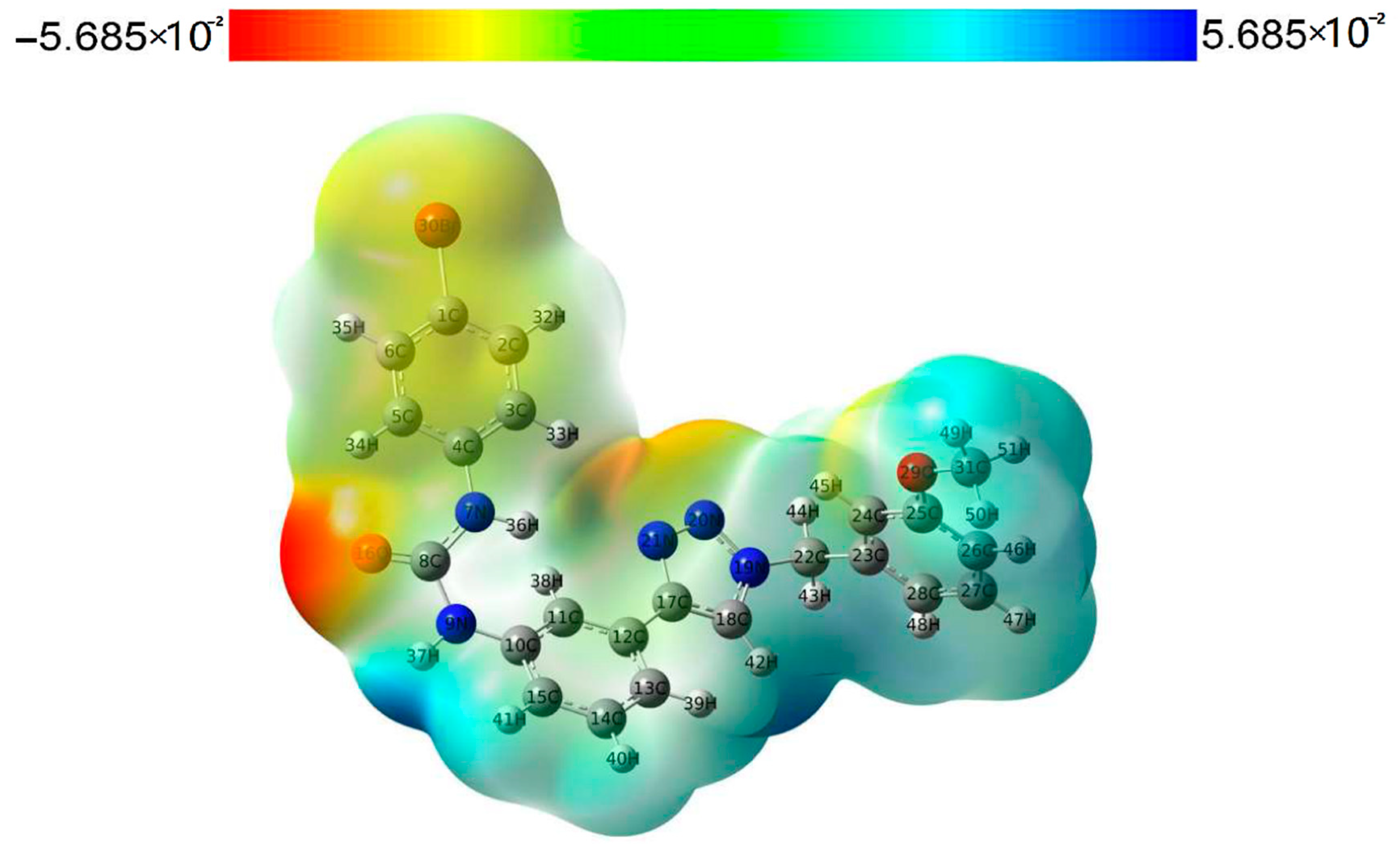
Molecular electrostatic potential (MEP) can help us obtain important information about the charge density, which is easy to ensure the sites of electrophilic nucleophilic reaction of muons, and also reflect the molecular interactions and physicochemical property characteristics [19,20]. We performed an MEP analysis to gain a better understanding of the electrophilic and nucleophilic reaction sites of compound 3a. The electrostatic potential surface map of compound 3a is shown in Figure 6. Different colors of the electrostatic potential surface represent the changing trend of the potential energy, and the increasing trend of the potential energy is red > yellow > green > blue. The negative regions (red and yellow) of the electrostatic potential map of the compound are correlated with electrophilic reactivity, and positive regions (blue) are related to nucleophilic reactivity. The highly negative region of compound 3a is located at O16 of the urea group, and the highly positive region is located at the NH group involved in the N9 of the urea group. The atoms in these regions may form a hydrogen bond interaction protein with IDO1, which is consistent with the docking results. In addition, the N20 and N21 of 1,2,3-triazole in the structure of compound 3a are located in the orange region with higher electron density, which can serve as the basic site for heme iron binding [21].
3. Materials and Methods
3.1. Materials and Chemistry
The 1,2,3-triazole derivatives were synthesized in-house by our research group. All reagents and solvents were obtained from a commercial source (Sinopharm, Beijing, China) and used without further treatment. 1H NMR and 13C NMR spectra were recorded in DMSO-d6 solution with a Bruker 600 spectrometer (Bruker, Karlsruhe, Germany). Chemical shifts (d) were recorded in parts per million. Tetramethylsilane (Acros, Organics, Brussels, Belgium) was used as an internal reference, and coupling constants were expressed in hertz. High-resolution mass spectra (HRMS) measurements were carried out using a Bruker Micro ToF II mass spectrometer (Bruker, Karlsruhe, Germany).
Hela cell line and DMEM mediumand fetal bovine serum were purchased from ATCC (Manassas, VA, USA) commercially, and Recombinant Human IFN-γ was purchased from R&D systems (Emeryville, CA, USA) commercially. The 3.05 Ntrichloroacetic acid, 4-(dimethylamino) benzaldehyde, and acetic acid were purchased from Sigma Aldrich (St. Louis, MO, USA).
3.2. General Procedure for Preparation of Compound 2a (The Method Is Suitable for 2b, 4a, 4b, 6a and 6b)
To a solution of 4-bromophenyl isocyanate (2.0 g, 0.01 mol) in CH2Cl2 (100 mL), 3-aminophenylacetylene (1.2 g, 0.01 mol) was addedin one portion. After stirring at room temperature for 2.5 h, the mixture was concentrated, and the residue was purified by column chromatography on silica gel (eluent:PE:EA = 2:1) to make compound 2a a pale yellow solid.
3.3. General Procedure for Preparation of Compound 3a
The reaction reagents were compound 2a (0.32 g, 1.0 mmol) and1-(azidomethyl)-3-methoxybenzene (0.2 g, 1.2 mmol), which were added to 15 mL of mixed solvent (water:tert-butanol = 2:1). The reaction was performed in copper sulfate pentahydrate (0.1 mmol) and sodium ascorbate (0.2 mmol) at 80 °C. The reaction status was monitored by TLC, and after completion of the reaction, the mixture was extracted through dichloromethane (15 mL × 3). The organic phase was combined and washed successively with water and brine, then dried over sodium sulfate and concentrated in vacuo. The desired compound 3a was isolated by column chromatography (CH2Cl2/MeOH = 20:1).
1-(4-bromophenyl)-3-(3-(1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)urea (3a). White solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2 [M+H]+478.0879, found 478.0876. 1H NMR (600 MHz, DMSO-d6) δ 8.82 (s, 2H), 8.60 (s, 1H), 8.03 (s, 1H), 7.45–7.42 (m, 5H), 7.36–7.28 (m, 3H), 6.96 (t, J = 2.4 Hz, 1H), 6.92 (d, J = 4.2 Hz, 1H), 6.90 (d, J = 3.6 Hz, 1H), 5.60 (s, 2H), 3.75 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.47, 152.39, 146.61, 140.05, 139.10, 137.45, 131.51, 129.97, 129.38, 121.60, 120.19, 120.01, 119.05, 117.85, 114.92, 113.77, 133.50, 113.25, 55.12, 55.92.
1-(3-(1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-methoxyphenyl)urea (3b). White solid. HR MS (ESI) m/z: calcd for C24H24N5O3 [M+H]+430.1879, found 430.1871. 1H NMR (600 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.60 (s, 1H), 8.48 (s, 1H), 8.03 (t, J = 1.8 Hz, 1H), 7.41 (dt, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.39–7.37 (m, 3H), 7.34-7.29 (m, 2H), 6.97 (t, J = 1.8 Hz, 1H), 6.92 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 2H), 6.89-6.87 (m, 2H), 5.61 (s, 2H), 3.75 (s, 3H), 3.72 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.95,155.00, 153.20, 147.17, 140.93, 137.94, 133.13, 131.62, 130.44, 129.81, 122.04, 120.58, 120.49, 119.18, 118.11, 155.18, 144.47, 114.24, 113.97, 55.65, 55.59, 53.41.
1-(4-fluorophenyl)-3-(3-(1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)urea (3c). Gray solid. HR MS (ESI) m/z: calcd for C23H21FN5O2 [M+H]+418.1679, found 418.1684. 1H NMR (600 MHz, DMSO-d6) δ 8.77 (s, 1H), 8.70 (s, 1H), 8.60 (s, 1H), 8.02 (t, J = 1.8 Hz, 1H), 7.50–7.46 (m, 2H), 7.42 (dt, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 7.37 (dt, J1 = 9.0 Hz, J2 = 1.2 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.12 (m, 2H), 6.96 (t, J = 1.8 Hz, 1H), 6.91 (dd, J1 = 8.4 Hz, J2 = 3.0 Hz, 2H), 5.60 (s, 2H), 3.75 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.47, 156.58, 152.63, 146.64, 140.22, 137.46, 131.16, 129.96, 129.36, 121.58, 120.07, 120.01, 118.91, 117.78, 115.34, 115.19, 114.85, 113.76, 113.49, 55.12, 52.93.
1-(3-(1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-phenylthiourea (3d). White solid. HR MS (ESI) m/z: calcd for C23H22N5OS[M+H]+416.1545, found 416.1546. 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.82 (s, 1H), 7.79 (s, 1H), 7.68 (s, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.47–7.41 (m, 4H), 7.38 (t, J = 7.2 Hz, 2H), 7.30 (t, J = 8.0 Hz, 2H), 6.91–6.88 (m, 2H), 6.82 (t, J = 1.6 Hz, 1H), 5.53 (s, 2H), 3.79 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 179.12, 159.31, 146.35, 137.09, 135.89, 135.00, 131.01, 129.44, 128.95, 128.89, 126.51, 124.57, 123.89, 123.13, 121.23, 119.43, 119.11, 113.46, 112.88, 54.47, 53.42.
1-(3-(1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-bromophenyl)urea (3e). White solid. HR MS (ESI) m/z: calcd for C22H18Br2N5O [M+H]+525.9878, found 525.9868. 1H NMR (600 MHz, DMSO-d6) δ 8.84 (s, 2H), 8.66 (s, 1H), 8.04 (t, J = 1.8 Hz, 1H), 7.61 (s, 1H), 7.57–7.55 (m, 1H), 7.46 (s, 3H), 7.44 (dt, J1 = 7.2 Hz, J2 = 1.8 Hz, 1H), 7.37–7.36 (m, 3H), 6.87 (s, 1H), 6.65 (s, 1H), 5.66 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.87, 151.94, 147.16, 139.66, 139.12, 131.99, 131.55, 131.51, 131.23, 128.51, 127.56, 125.39, 122.35, 122.24, 120.65, 118.37, 115.38, 113.73, 52.63, 34.86, 30.89, 21.52.
1-(3-(1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-methoxyphenyl)urea (3f). Brown solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2 [M+H]+478.0879, found 478.0875. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.64 (s, 1H), 8.49 (s, 1H), 8.03 (s, 1H), 7.61 (s, 1H), 7.56–7.55 (m, 1H), 7.38 (s, 1H), 7.36 (d, J = 4.2 Hz, 4H), 7.33 (d, J = 7.8 Hz, 1H), 6.87 (d, J = 9.0 Hz, 2H), 3.72 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 154.97, 153.19, 147.24, 140.95, 139.13, 131.55, 131.51, 131.23, 129.83, 127.56, 122.35, 122.21, 120.55, 119.16, 118.14, 115.15, 114.45, 55.64, 52.63, 30.89.
1-(3-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-bromophenyl)urea (3g). Gray solid. HR MS (ESI) m/z: calcd for C22H18Br2N5O [M+H]+525.9878, found 525.9885. 1H NMR (600 MHz, DMSO-d6) δ 8.84 (d, J = 6.0 Hz, 2H), 8.58 (s, 1H), 8.03 (t, J = 1.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.46 (s, 4H), 7.44 (d, J = 1.8 Hz, 1H), 7.44-7.42 (m, 1H), 7.40 (dd, J1 = 6.6 Hz, J2 = 1.8 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.33 (dd, J1 = 7.8 Hz, J2 = 1.8 Hz, 1H), 7.22 (dd, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 5.74 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.88, 146.92, 140.54, 138.57, 135.34, 133.39, 131.99, 131.57, 130.90, 129.88, 128.82, 123.31, 122.52, 120.71, 120.66, 119.59, 118.38, 115.41, 113.73, 53.56.
1-(3-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-fluorophenyl)urea (3h). Gray solid. HR MS (ESI) m/z: calcd for C22H18BrFN5O [M+H]+466.0679, found 466.0673. 1H NMR (600 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.90 (s, 1H), 8.59 (s, 1H), 8.03 (t, J = 1.8 Hz, 1H), 7.71 (dd, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.48 (dd, J1 = 9.0 Hz, J2 = 4.8 Hz, 2H), 7.43 (t, J = 6.6 Hz, 2H), 7.40 (d, J = 7.8 Hz, 1H), 7.35-7.33 (m, 2H), 7.22 (dd, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.13 (t, J = 8.4 Hz, 2H), 5.74 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 153.15, 146.96, 140.77, 135.36, 133.39, 131.54, 130.88, 129.84, 128.82, 125.39, 123.30, 122.49, 120.43, 120.38, 119.40, 118.25, 115.82, 115.68, 115.28, 53.55.
1-(3-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-methoxyphenyl)urea (3i). White solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2 [M+H]+478.0879, found 478.0882. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.57 (s, 1H), 8.48 (s, 1H), 8.02 (t, J = 1.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.44–7.41 (m, 2H), 7.40–7.36 (m, 3H), 7.33 (t, J = 7.8 Hz, 2H), 7.22 (dd, J1 = 7.2 Hz, J2 = 1.8 Hz, 1H), 6.89–6.86 (m, 2H), 5.74 (s, 2H), 3.71 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 154.98, 153.19, 146.99, 140.93, 139.67, 135.36, 133.39, 133.11, 131.51, 130.89, 129.83, 128.82, 125.39, 123.30, 122.56, 119.56, 118.15, 115.18, 114.45, 55.64, 53.56.
1-(4-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-methoxyphenyl)urea (5a). White solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2 [M+H]+478.0879, found 478.0866. 1H NMR (600 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.49 (d, J = 11.4 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 9.0 Hz, 2H), 7.43 (t, J = 7.2 Hz, 1H), 7.36 (d, J = 9.0 Hz, 2H), 7.34–7.31 (m, 1H), 7.21 (d, J = 7.2 Hz, 1H), 6.87 (d, J = 9.0 Hz, 2H), 5.72 (s, 2H), 3.72 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.00, 153.11, 147.01, 140.20, 135.36, 133.39, 133.10, 130.89, 128.81, 126.27, 124.46, 123.31, 121.50, 120.56, 118.71, 114.47, 55.65, 53.54.
1-(4-(1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-methoxyphenyl)urea (5b). Brown solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2[M+H]+478.0879, found 478.0887. 1H NMR (600 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.55 (s, 1H), 8.49 (s, 1H), 7.74 (d, J = 9.0 Hz, 2H), 7.59 (s, 1H), 7.55 (d, J = 7.2 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 2.4 Hz, 2H), 7.36–7.35 (m, 2H), 6.87 (d, J = 9.0 Hz, 2H), 5.65 (s, 2H), 3.71 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.00, 153.11, 147.25, 140.20, 139.16, 133.09, 131.53, 131.50, 131.17, 127.51, 126.25, 124.46, 122.35, 121.26, 120.56, 118.72, 114.47, 55.65, 52.61.
1-(4-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-bromophenyl)urea (5c). White solid. HR MS (ESI) m/z: calcd for C22H18Br2N5O [M+H]+525.9878, found 525.9870. 1H NMR (600 MHz, DMSO-d6) δ 8.83 (d, J = 13.2 Hz, 2H), 8.50 (s, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.71 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.46–7.44 (m, 4H), 7.42 (dd, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 7.33 (td, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.22 (dd, J1 = 7.8 Hz, J2 = 1.8 Hz, 1H), 5.72 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.79, 146.94, 139.77, 139.56, 135.34, 133.39, 131.99, 130.90, 128.81, 126.30, 124.85, 123.32, 121.60, 120.65, 118.96, 113.75, 53.55.
1-(4-(1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-bromophenyl)urea (5d). White solid. HR MS (ESI) m/z: calcd for C22H18Br2N5O [M+H]+525.9878, found 525.9881. 1H NMR (600 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.83 (s, 1H), 8.56 (s, 1H), 7.76 (d, J = 9.0 Hz, 2H), 7.59 (s, 1H), 7.56 (dt, J1 = 7.2 Hz, J2 = 1.8 Hz, 1H), 7.53 (d, J = 9.0 Hz, 2H), 7.45 (s, 4H), 7.38–7.34 (m, 2H), 5.65 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.79, 147.18, 139.79, 139.56, 139.14, 131.99, 131.53, 131.50, 131.18, 127.52, 126.27, 124.84, 122.35, 121.36, 120.65, 118.97, 113.74, 52.62.
1-(4-bromophenyl)-3-(4-(1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)phenyl)urea (5e). White solid. HR MS (ESI) m/z: calcd for C23H21BrN5O2 [M+H]+478.0879, found 478.0886. 1H NMR (600 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.84 (s, 1H), 8.53 (s, 1H), 7.76 (d, J = 9.0 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.45 (s, 4H), 7.31 (t, J = 7.8 Hz, 1H), 6.95 (s, 1H), 6.93–6.89 (m, 2H), 5.59 (s, 2H), 3.75 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.95, 152.80, 147.10, 139.74, 139.58, 137.96, 131.99, 130.44, 126.24, 124.94, 121.21, 120.64, 120.45, 118.97, 114.20, 113.96, 113.73, 55.60, 53.39.
1-(4-(1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)-3-(4-chlorophenyl)urea (5f). White solid. HR MS (ESI) m/z: calcd for C22H18BrClN5O [M+H]+482.0383, found 482.0374. 1H NMR (600 MHz, DMSO-d6) δ 8.86 (d, J = 13.2 Hz, 2H), 8.52 (s, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.72 (dd, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.52-7.49(m, 2H), 7.44 (td, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.35-7.32(m, 3H), 7.22 (dd, J1 = 7.8 Hz, J2 = 1.8 Hz, 1H), 5.73 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 152.82, 146.93, 139.79, 139.68, 139.12, 135.35, 133.39, 130.89, 129.12, 128.82, 126.29, 125.85, 125.40, 124.81, 123.31, 121.61, 120.22, 118.93, 53.54, 34.87, 30.88, 21.52.
1-((1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-methoxyphenyl)urea (7a). Gray solid. HR MS (ESI) m/z: calcd for C18H19BrN5O2[M+H]+416.0722, found 416.0714. 1H NMR (600 MHz, DMSO-d6) δ 8.34 (s, 1H), 7.97 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.40 (td, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 7.31 (td, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 7.28 (d, J = 8.4 Hz, 2H), 7.14 (dd, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 6.81 (d, J = 9.0 Hz, 2H), 6.47 (t, J = 5.4 Hz, 1H), 5.66 (s, 2H), 4.32 (d, J = 5.4Hz, 2H), 3.69 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.78, 154.47, 135.51, 133.94, 133.37, 130.85, 130.83, 128.74, 123.67, 123.30, 119.94, 114.35, 55.61, 53.29, 35.36.
1-((1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-methoxyphenyl)urea (7b). Gray solid. HR MS (ESI) m/z: calcd for C18H19BrN5O2[M+H]+416.0722, found 416.0729. 1H NMR (600 MHz, DMSO-d6) δ 8.34 (s, 1H), 8.08 (s, 1H), 7.53 (d, J = 9.6 Hz, 2H), 7.34 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 9.0 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 6.47 (s, 1H), 5.59 (s, 2H), 4.31 (s, 2H), 3.69 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.85, 154.47, 139.25, 133.94, 131.48, 131.43, 131.20, 127.56, 123.74, 122.28, 119.94, 114.34, 55.61, 52.37, 35.43.
1-((1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-methoxyphenyl)urea (7c). White solid. HR MS (ESI) m/z: calcd for C19H22N5O3 [M+H]+368.1723, found 368.1731. 1H NMR (600 MHz, DMSO-d6) δ 8.34 (s, 1H), 8.03 (s, 1H), 7.29-7.26 (m, 3H), 6.89 (d, J = 6.0 Hz, 2H), 6.86 (d, J = 7.2 Hz, 1H), 6.81 (d, J = 9.0 Hz, 2H), 6.46 (s, 1H), 5.53 (s, 2H), 4.31 (s, 2H), 3.73 (s, 3H), 3.69 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.88, 155.84, 154.47, 138.02, 133.93, 130.36, 120.50, 119.93, 114.34, 114.24, 113.87, 55.60, 55.56, 53.16, 35.42.
1-((1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-bromophenyl)urea (7d). White solid. HR MS (ESI) m/z: calcd for C17H16Br2N5O [M+H]+463.9722, found 463.9729. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.99 (s, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.41–7.35 (m, 5H), 7.31 (td, J1 = 7.8 Hz, J2 = 1.2 Hz, 1H), 7.15 (d, J = 6.6 Hz, 1H), 6.64 (t, J = 4.8 Hz, 1H), 5.66 (s, 2H), 4.34 (d, J = 5.4 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 155.36, 140.27, 135.49, 133.37, 131.83, 130.86, 130.83, 128.74, 123.76, 123.30, 120.08, 112.90, 53.31, 35.35.
1-((1-(3-bromobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-bromophenyl)urea (7e). White solid. HR MS (ESI) m/z: calcd for C17H16Br2N5O [M+H]+463.9722, found 463.9727. 1H NMR (600 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.06 (s, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.39–7.35 (m, 4H), 7.33 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 7.2 Hz, 1H), 6.64 (s, 1H), 5.59 (s, 2H), 4.33 (d, J = 4.8 Hz, 2H). 13C NMR (150MHz, DMSO-d6) δ 155.36, 140.27, 139.26, 131.82, 131.48, 131.43, 131.19, 127.56, 123.50, 122.28, 120.08, 112.90, 52.35, 35.36.
1-(4-bromophenyl)-3-((1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)urea (7f). Brown solid. HR MS (ESI) m/z: calcd for C18H19BrN5O2[M+H]+416.0722, found 416.0715. 1H NMR (600 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.00 (s, 1H), 7.39–7.36 (m, 4H), 7.30–7.25 (m, 1H), 6.90–6.88 (m, 2H), 6.86 (d, J = 7.8 Hz, 1H), 6.63 (t, J = 5.4 Hz, 1H), 5.53 (s, 2H), 4.32 (d, J = 5.4Hz, 2H), 3.73 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 159.89, 155.34, 140.27, 138.06, 131.83, 130.36, 123.27, 120.50, 120.07, 114.24, 113.86, 112.89, 55.56, 53.11, 35.35.
1H NMR spectrum of compounds 3a–3i, 5a–5f and 7a–7f are shown in Figures S1–S21.
3.4. IDO1 Enzymatic Inhibition Assay
To demonstrate the inhibitory effects of the designed compounds against IDO1, the Hela cell density was adjusted using DMEM complete medium and subsequently seeded into 96-well cell culture plates at 100 μL per well for a total of 50,000 cells, and then incubated at 37 °C, 5% CO2 overnight. The next day, 100 μL of various concentrations of the compounds were diluted in a medium containing 100 ng/mL human IFNγ, added onto the 96-well plate, then incubated for 18 h. On the third day, 140 μL of the medium was removed from a new 96-well plate and 10 μL of 6.1 N TCA was added to precipitate the protein at 50 °C; for 30 min. The sediment was centrifuged at 2500 rpm for 10 min. Then, the supernate was transferred to another 96-well plate and mixed with 100 μL (2% (w/v)) of 4-(dimethylamino) benzaldehyde in acetic acid. The plate was incubated at room temperature for 10 min, and the yellow color derived from the kynurenine was recorded by measuring absorbance at 480 nm using a microplate reader (PE, Envision; Perkinelmer & Co.: Wellesley, MA, USA, 2019). Graphs of inhibition curves with IC50 values were generated using Prism 6.0 [2,22].
3.5. Molecular Docking
Docking experiments were performed by using Schrödinger (LLC 2015; Software for Technical Computation; Schrödinger Inc.: New York, NY, USA, 2015), and 4PK5 with Amg-1 as the co-crystal ligand was chosen as the complex system. Protein was processed and optimized by the Protein Preparation Wizard, which included hydrogenation of protein, assigned bond orders, creation of zero-order bonds with metals, creation of disulfide bonds, removal of water molecules larger than 5 Å from het groups, structural optimization, and energy minimization. The ligands were constructed by ChemDraw (version 16.0; Software for Technical Computation; Perkinelmer & Co.: Wellesley, MA, USA, 2016) and optimized by the LigPrep module under the condition of pH 7.0 ± 2.0 for protonation and generation of stereoisomers. Molecular lattices were generated centered on Amg-1, while metal coordination constraints centered on heme iron were generated; docking was performed using SP precision. The 3D binding patterns of compounds to IDO1 were visualized by PyMOL (version 2.4.0; Software for Technical Computation; Delano Scientific LLC: Silicon Valley, CA, USA, 2012).
3.6. Quantum Mechanical Studies (QM)
To better understand the molecular properties, we performed density functional theory (DFT) studies on compounds 3a, 5e and 7f based on quantum chemistry [23]. DFT is used to study the electronic structure of multi-electron systems, which has been widely used to calculate the electronic properties of molecules. The DFT calculation was implemented by Gaussian09 software (Software for Technical Computation; Gaussian Inc.: PIT, USA, 2017). Geometry optimization of compounds wasperformed by using the B3LYP/6–31G (d,p) basis sets [24]. The B3LYP/6–311G (d,p) basis sets were applied for the frontier orbital and molecular electrostatic potential (MEP)analysis. The highest occupied molecular orbital (HOMO) energy, the lowest unoccupied molecular orbital (LUMO) energy, and the energy gap () parameters were investigated.
4. Conclusions
In this study, three series of compounds containing a urea structure and 1,2,3-triazole structure were designed, among which, the compound 3 series exhibited better inhibitory effects on IDO1, especially compound 3a (IC50 = 0.75 μM). Molecular docking and quantum mechanical studies showed that compound 3a was more chemically reactive than 5e and 7f with the same substituents, further demonstrating the potential activity of compound 3a. Therefore, we will optimize the compound 3 series and carry out experimental studies at the cellular and animal levels in the future.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/ph15111316/s1, Figure S1: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3a; Figure S2: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3b; Figure S3: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3c; Figure S4: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3d; Figure S5: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3e; Figure S6: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3f; Figure S7: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3g; Figure S8: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3h; Figure S9: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 3i; Figure S10: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5a; Figure S11: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5b; Figure S12: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5c; Figure S13: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5d; Figure S14: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5e; Figure S15: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 5f; Figure S16: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7a; Figure S17: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7b; Figure S18: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7c; Figure S19: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7d; Figure S20: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7e; Figure S21: 1H NMR spectrum (600 MHz, DMSO-d6) of compound 7f.
Author Contributions
Conceptualization, J.Y.; methodology, X.H. and J.Z.; software, X.G.; validation, L.M.; data curation, J.Z.; writing—original draft preparation, X.H.; writing—review and editing, J.Y.; visualization, X.G.; funding acquisition, J.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Scientific and Technological Project of Henan Province (no. 192102310142).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Macan, A.M.; Harej, A.; Cazin, I.; Klobucar, M.; Stepanic, V.; Pavelic, K.; Pavelic, S.K.; Schols, D.; Snoeck, R.; Andrei, G.; et al. Antitumor and antiviral activities of 4-substituted 1,2,3-triazolyl-2,3-dibenzyl-L-ascorbic acid derivatives. Eur. J. Med. Chem. 2019, 184, 111739. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.F.; Wang, Y.W.; Zhao, J.; Xu, G.Q.; Yao, X.J.; Li, Y.M. Discovery of Icotinib-1,2,3-Triazole Derivatives as IDO1 Inhibitors. Front. Pharmacol. 2020, 11, 579024. [Google Scholar] [CrossRef] [PubMed]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Zhou, Y.; Wang, Q.; Wang, Y.; Tian, C.; Wang, T.; Huang, L.; Nan, J.; Li, L.; Yang, S. Discovery and structure-activity relationship studies of 1-aryl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione derivatives as potent dual inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) and trytophan 2,3-dioxygenase (TDO). Eur. J. Med. Chem. 2020, 207, 112703. [Google Scholar] [CrossRef] [PubMed]
- Partyka, A.; Chlon-Rzepa, G.; Wasik, A.; Jastrzebska-Wiesek, M.; Bucki, A.; Kolaczkowski, M.; Satala, G.; Bojarski, A.J.; Wesolowska, A. Antidepressant- and anxiolytic-like activity of 7-phenylpiperazinylalkyl-1,3-dimethyl-purine-2,6-dione derivatives with diversified 5-HT(1)A receptor functional profile. Bioorg. Med. Chem. 2015, 23, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, U.F.; Reynaud, A.; Majjigapu, S.R.; Vogel, P.; Pojer, F.; Zoete, V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2019, 62, 8784–8795. [Google Scholar] [CrossRef]
- Serafini, M.; Torre, E.; Aprile, S.; Grosso, E.D.; Gesù, A.; Griglio, A.; Colombo, G.; Travelli, C.; Paiella, S.; Adamo, A.; et al. Discovery of Highly Potent Benzimidazole Derivatives as Indoleamine 2,3-Dioxygenase-1 (IDO1) Inhibitors: From Structure-Based Virtual Screening to in Vivo Pharmacodynamic Activity. J. Med. Chem. 2020, 63, 3047–3065. [Google Scholar] [CrossRef]
- Soliman, H.; Antonia, S.; Sullivan, D.; Vanahanian, N.; Link, C. Overcoming tumor antigen anergy in human malignancies using the novel indeolamine 2,3-dioxygenase (IDO) enzyme inhibitor, 1-methyl-D-tryptophan (1MT). J. Clin. Oncol. 2009, 27, 3004. [Google Scholar] [CrossRef]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Jr, O.W.; Lew, N.L.; Liu, Y.; Lowrie, E.G.; Lazarus, J.M. The urea reduction ratio and serum albumin concentration as predictors of mortality in patients undergoing hemodialysis. N. Engl. J. Med. 1993, 329, 1001–1006. [Google Scholar]
- Kiser, J.J.; Burton, J.R.; Anderson, P.L.; Everson, G.T. Review and management of drug interactions with boceprevir and telaprevir. Hepatology 2012, 55, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2007, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L.P. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fling, S.P.; Gregerson, D.S. Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal. Biochem. 1986, 155, 83–88. [Google Scholar] [CrossRef]
- Nauck, M.; Frid, A.; Hermansen, K.; Thomsen, A.B.; During, M.; Shah, N.; Tankova, T.; Mitha, I.; Matthews, D.R. Long-term efficacy and safety comparison of liraglutide, glimepiride and placebo, all in combination with metformin in type 2 diabetes: 2-year results from the LEAD-2 study. Diabetes Obes. Metab. 2013, 15, 204–212. [Google Scholar] [CrossRef]
- Thanthiriwatte, K.S.; De Silva, K.N. Non-linear optical properties of novel fluorenyl derivatives-ab initio quantum chemical calculations. J. Mol. Struct. Theochem. 2002, 617, 169–175. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Celik, I.; Işık, A.; Ahmad, I.; Patel, H.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, molecular modeling, DFT, ADME and biological evaluation studies of some new 1, 3, 4-oxadiazole linked benzimidazoles as anticancer agents and aromatase inhibitors. J. Biomol. Struct. Dyn. 2022, 6, 1–15. [Google Scholar] [CrossRef]
- Sureshkumar, B.; Mary, Y.S.; Panicker, C.Y.; Suma, S.; Armakovic, S.; Armakovic, S.J.; Alsenoy, C.V.; Narayana, B. Quinolinederivatives as possible lead compounds for anti-malarial drugs: Spectroscopic, DFT and MD study. Arab. J. Chem. 2020, 13, 632–648. [Google Scholar] [CrossRef]
- Sevvanthi, S.; Muthu, S.; Raja, M.; Aayisha, S.; Janani, S. PES, molecular structure, spectroscopic (FT-IR, FT-Raman), electronic (UV-Vis, HOMO-LUMO),quantum chemical and biological (docking) studies on a potent membrane permeable inhibitor:dibenzoxepine derivative. Heliyon 2020, 6, 4724. [Google Scholar] [CrossRef]
- Gaspari, P.; Banerjee, T.; Malachowski, W.P.; Muller, A.J.; Prendergast, G.C.; DuHadaway, J.; Bennett, S.; Donovan, A.M. Structure-Activity Study of Brassinin Derivatives as Indoleamine 2,3-Dioxygenase Inhibitors. J. Med. Chem. 2006, 49, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.X.; Gong, X.Q.; Mao, L.F.; Sun, G.; Yang, J.X. Design, synthesis and biological evaluationof erlotinib-based IDO1 inhibitors. Front. Pharmacol. 2022, 10, 940704. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Tao, Y.L.; Qu, H.Y.; Wang, C.H.; Yan, F.; Gao, X.J.; Zhang, M.L. Exploration of plant-derived natural polyphenols toward COVID-19 main protease inhibitors: DFT, molecular docking approach, and molecular dynamics simulations. RSC Adv. 2022, 12, 5357–5368. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Gobinath, E. Experimental and theoretical spectroscopic studies, HOMO–LUMO, NBO and NLMO analysis of3,5-dibromo-2,6-dimethoxy pyridine. Spectrochim. Acta A 2012, 97, 215–222. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structures of four IDO1 inhibitors.

Figure 2.
Pocket surface map of the crystal structure of IDO1 complexed with Amg-1 (PDB code: 4PK5). Purple represents amino acids contained in pocket A; green represents amino acids contained in pocket B.
Figure 2.
Pocket surface map of the crystal structure of IDO1 complexed with Amg-1 (PDB code: 4PK5). Purple represents amino acids contained in pocket A; green represents amino acids contained in pocket B.

Figure 3.
The reaction routes to compounds 3a–3i, 5a–5f, and 7a–7f.

Figure 4.
The binding modes of compounds 3a (A), 5e (B) and 7f (C).

Figure 5.
Frontier molecular orbitals of compounds 3a (A), 5e (B) and 7f (C).

Figure 6.
MEP surfaces of compound 3a.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
R-group of compounds 3a–3i, 5a–5f, and 7a–7f.
| Compound | X | R1 | R2 | R3 | Yield (%) |
|---|---|---|---|---|---|
| 3a | O | Br | H | OCH3 | 73.9 |
| 3b | O | OCH3 | H | OCH3 | 55.1 |
| 3c | O | F | H | OCH3 | 80.8 |
| 3d | S | H | H | OCH3 | 69.2 |
| 3e | O | Br | H | Br | 85.9 |
| 3f | O | OCH3 | H | Br | 77.4 |
| 3g | O | Br | Br | H | 86.6 |
| 3h | O | F | Br | H | 63.8 |
| 3i | O | OCH3 | Br | H | 49.1 |
| 5a | O | OCH3 | Br | H | 41.7 |
| 5b | O | OCH3 | H | Br | 87.2 |
| 5c | O | Br | Br | H | 39.4 |
| 5d | O | Br | H | Br | 33.4 |
| 5e | O | Br | H | OCH3 | 40.2 |
| 5f | O | Cl | Br | H | 76.4 |
| 7a | O | OCH3 | Br | H | 36.6 |
| 7b | O | OCH3 | H | Br | 43.9 |
| 7c | O | OCH3 | H | OCH3 | 39.8 |
| 7d | O | Br | Br | H | 43.2 |
| 7e | O | Br | H | Br | 86.9 |
| 7f | O | Br | H | OCH3 | 45.4 |
Table 2.
IDO1 inhibitory activities of designed derivatives.
| Compd No. | IC50 (μM) | Compd No. | IC50 (μM) | Compd No. | IC50 (μM) |
|---|---|---|---|---|---|
| IDO1 | IDO1 | IDO1 | |||
| 3a | 0.75 ± 0.27 | 5a | >10 | 7a | >10 |
| 3b | 3.46 ± 0.71 | 5b | 7.43 ± 1.05 | 7b | 3.97 ± 0.92 |
| 3c | 6.21 ± 0.89 | 5c | >10 | 7c | 7.10 ± 1.48 |
| 3d | 4.13 ± 0.42 | 5d | >10 | 7d | >10 |
| 3e | 8.16 ± 1.12 | 5e | >10 | 7e | >10 |
| 3f | 5.93 ± 0.97 | 5f | >10 | 7f | >10 |
| 3g | 2.59 ± 0.13 | ||||
| 3h | 2.58 ± 0.59 | ||||
| 3i | 0.80 ± 0.37 |
IC50 values were fitted from single-point inhibition curves, and two parallel experiments were performed for each compound. IC50 values were calculated using Graph Pad Prism 6.0 software. These results are reported as the averages ± SD.
Table 3.
Molecular frontier orbital parameters of compounds 3a, 5e and 7f.
| Compd No. | HOMO Energy (eV) | LUMO Energy (eV) | |
|---|---|---|---|
| 3a | −5.8395 | −1.2633 | 4.5762 |
| 5e | −5.9690 | −1.2998 | 4.6692 |
| 7f | −5.8782 | −0.8868 | 4.9914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hou, X.; Gong, X.; Mao, L.; Zhao, J.; Yang, J. Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors. Pharmaceuticals 2022, 15, 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15111316
AMA Style
Hou X, Gong X, Mao L, Zhao J, Yang J. Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors. Pharmaceuticals. 2022; 15(11):1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15111316
Chicago/Turabian StyleHou, Xixi, Xiaoqing Gong, Longfei Mao, Jie Zhao, and Jianxue Yang. 2022. "Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors" Pharmaceuticals 15, no. 11: 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15111316
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.