
SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies


Abstract
:1. Introduction
2. Valproic Acid (VPA)
2.1. VPA and Autism Spectrum Disorder (ASD)
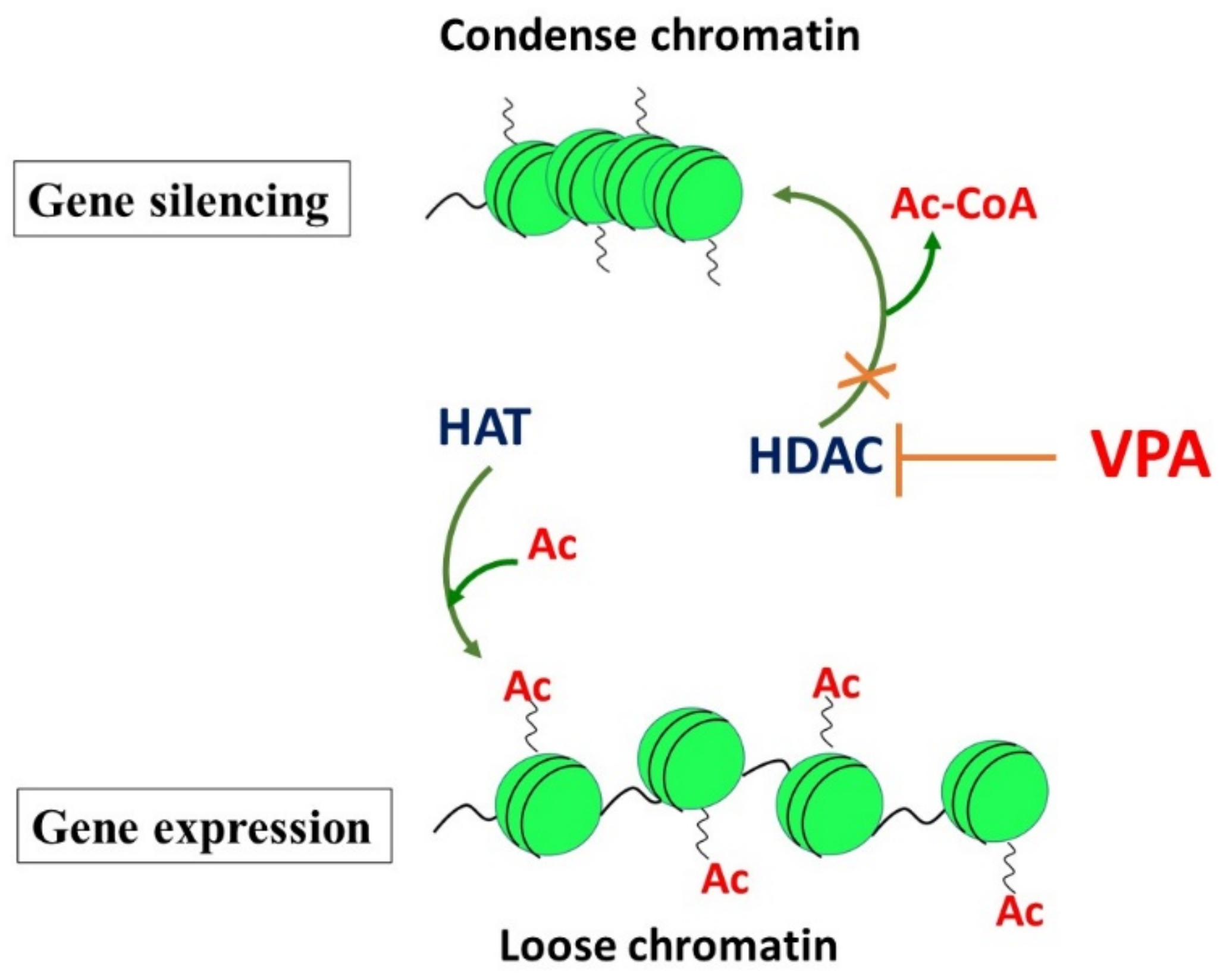
2.2. Inhibition of Histone Deacetylases (HDACs) by VPA: The Effects on the Epigenome
2.3. Changes in Gene Expression in Offspring Induced by VPA Exposure during the Prenatal or Early Postnatal Periods
2.4. In Summary
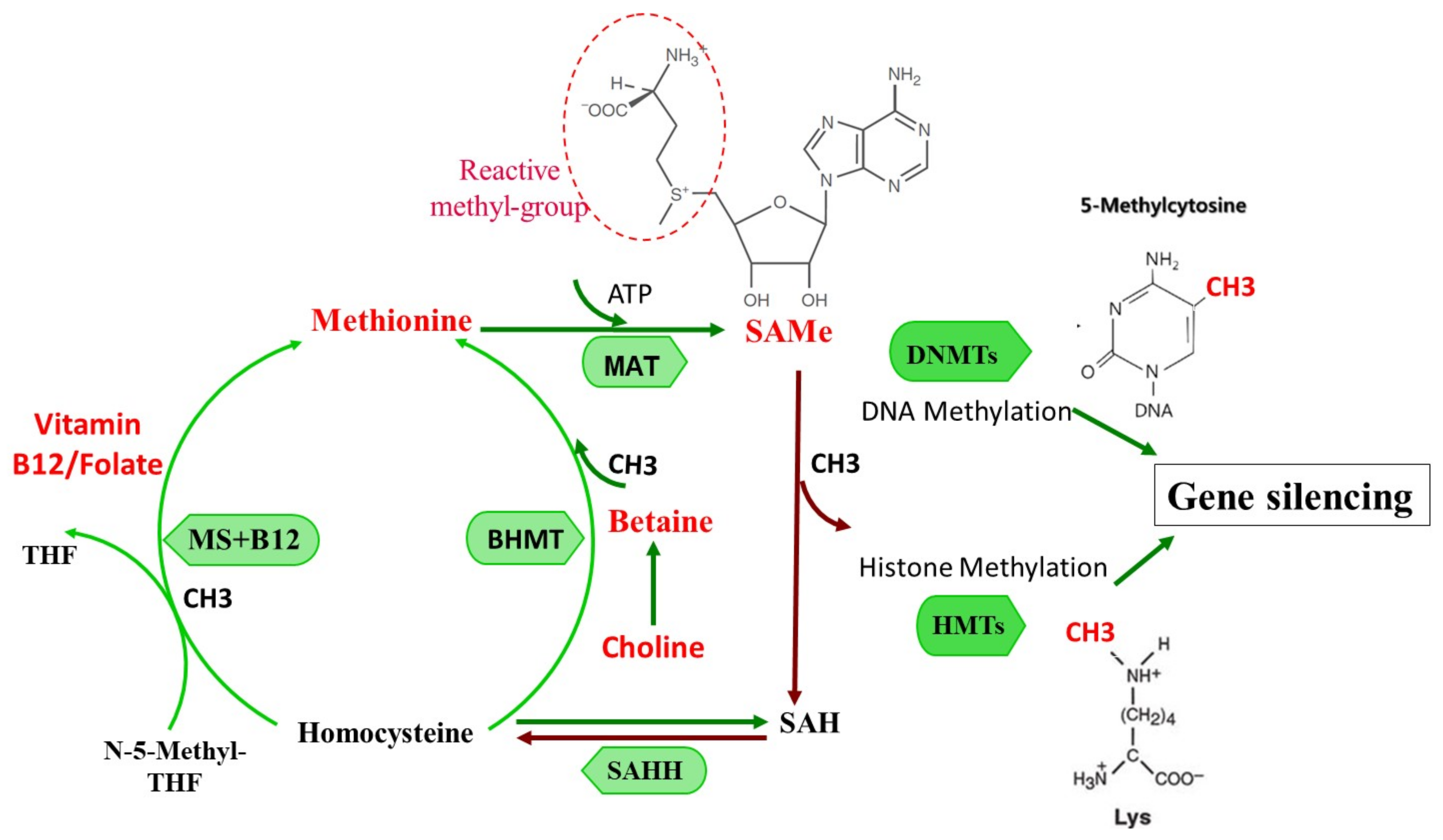
3. S-adenosylmethionine: The Principal Physiologic Methyl Donor
3.1. SAMe as an Epigenetic Modulator
3.2. SAMe Effects on the Embryo, Fetus, and Neonate: Its Role as an Epigenetic Modulator
3.3. SAMe and Its Possible Effects on the Pregnant Mother and Offspring
3.4. In Summary
4. Choline and Pregnancy
4.1. The Impact of Choline on Pregnancy and Offspring in Rodents
4.2. Choline and Prenatal Exposure to Alcohol: Experiments in Animals
4.3. Human Studies
4.4. Human Studies Assessing the Possible Effects of Gestational Choline on Offspring in Normal Pregnancies and in Pregnancies Following High Alcohol Ingestion
4.5. Early Postnatal Choline Treatment of Children with FASD
4.6. In Summary
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A(vy) | viable yellow agouti mice |
| ASD | Autism spectrum disorder |
| DNMTS | DNA methyltransferases |
| FASD | Fetal alcohol spectrum disorder |
| Flt1 | Fms related receptor tyrosine kinase 1 |
| GSH | Hepatic glutathione |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylases |
| ICM | Inner cell mass |
| MAT | Methionine adenosyltransferase |
| MDD | Major depressive disorder |
| MTHFR | Methylenetetrahydrofolate reductase |
| SAMe | S-adenosylmethionine |
| Sub | Submissive mice |
| TET | Ten–eleven translocation dioxygenases |
| VEGF | Vascular endothelial growth factor |
| VPA | Valproic acid |
References
- Szyf, M. Prospects for the development of epigenetic drugs for CNS conditions. Nat. Rev. Drug Discov. 2015, 14, 461–474. [Google Scholar] [CrossRef]
- Chiarella, J.; Schumann, L.; Pomares, F.B.; Frodl, T.; Tozzi, L.; Nemoda, Z.; Yu, P.; Szyf, M.; Khalid-Khan, S.; Booij, L. DNA methylation differences in stress-related genes, functional connectivity and gray matter volume in depressed and healthy adolescents. J. Affect. Disord. 2020, 271, 160–168. [Google Scholar] [CrossRef]
- Petropoulos, S.; Guillemin, C.; Ergaz, Z.; Dimov, S.; Suderman, M.; Weinstein-Fudim, L.; Ornoy, A.; Szyf, M. Gestational Diabetes Alters Offspring DNA Methylation Profiles in Human and Rat: Identification of Key Pathways Involved in Endocrine System Disorders, Insulin Signaling, Diabetes Signaling, and ILK Signaling. Endocrinology 2015, 156, 2222–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M. Perinatal stress and epigenetics. Handb. Clin. Neurol. 2021, 180, 125–148. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roost, M.S.; Slieker, R.C.; Bialecka, M.; van Iperen, L.; Gomes Fernandes, M.M.; He, N.; Suchiman, H.E.D.; Szuhai, K.; Carlotti, F.; de Koning, E.J.P.; et al. DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat. Commun. 2017, 8, 908. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Xie, W. Rebooting the Epigenomes during Mammalian Early Embryogenesis. Stem Cell Rep. 2020, 15, 1158–1175. [Google Scholar] [CrossRef] [PubMed]
- Reizel, Y.; Sabag, O.; Skversky, Y.; Spiro, A.; Steinberg, B.; Bernstein, D.; Wang, A.; Kieckhaefer, J.; Li, C.; Pikarsky, E.; et al. Postnatal DNA demethylation and its role in tissue maturation. Nat. Commun. 2018, 9, 2040. [Google Scholar] [CrossRef]
- Nagy, C.; Turecki, G. Sensitive periods in epigenetics: Bringing us closer to complex behavioral phenotypes. Epigenomics 2012, 4, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Burns, S.B.; Szyszkowicz, J.K.; Luheshi, G.N.; Lutz, P.E.; Turecki, G. Plasticity of the epigenome during early-life stress. Semin. Cell Dev. Biol. 2018, 77, 115–132. [Google Scholar] [CrossRef]
- Ornoy, A.; Ergaz, Z. Alcohol abuse in pregnant women: Effects on the fetus and newborn, mode of action and maternal treatment. Int. J. Environ. Res. Public Health 2010, 7, 364–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, P.J.; Naninck, E.F.; van Goudoever, J.B.; Fitzsimons, C.; Joels, M.; Korosi, A. Perinatal programming of adult hippocampal structure and function; emerging roles of stress, nutrition and epigenetics. Trends Neurosci. 2013, 36, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Maire, C.L.; Bilenky, M.; Carles, A.; Heravi-Moussavi, A.; Hong, C.; Tam, A.; Kamoh, B.; Cho, S.; Cheung, D.; et al. Epigenomic programming in early fetal brain development. Epigenomics 2020, 12, 1053–1070. [Google Scholar] [CrossRef]
- Bottom, R.T.; Abbott, C.W., 3rd; Huffman, K.J. Rescue of ethanol-induced FASD-like phenotypes via prenatal co-administration of choline. Neuropharmacology 2020, 168, 107990. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Fink, B.A.; Fuglestad, A.J.; Eckerle, J.K.; Boys, C.J.; Sandness, K.E.; Radke, J.P.; Miller, N.C.; Lindgren, C.; Brearley, A.M.; et al. Four-year follow-up of a randomized controlled trial of choline for neurodevelopment in fetal alcohol spectrum disorder. J. Neurodev. Disord. 2020, 12, 9. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicol. Teratol. 2019, 71, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Weinstein-Fudim, L.; Ergaz, Z.; Turgeman, G.; Yanai, J.; Szyf, M.; Ornoy, A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? Int. J. Mol. Sci. 2019, 20, 5278. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.N.K.; Kichenadasse, G.; Morel, K.L.; Lavranos, T.C.; Klebe, S.; Lower, K.M.; Ormsby, R.J.; Elliot, D.J.; Sykes, P.J. The Combination of Metformin and Valproic Acid Has a Greater Anti-tumoral Effect on Prostate Cancer Growth In Vivo than Either Drug Alone. Vivo 2019, 33, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Kaaja, E.; Kaaja, R.; Hiilesmaa, V. Major malformations in offspring of women with epilepsy. Neurology 2003, 60, 575–579. [Google Scholar] [CrossRef]
- Vajda, F.J.; Hitchcock, A.; Graham, J.; O’Brien, T.; Lander, C.; Eadie, M. The Australian Register of Antiepileptic Drugs in Pregnancy: The first 1002 pregnancies. Aust. N. Z. J. Obstet. Gynaecol. 2007, 47, 468–474. [Google Scholar] [CrossRef]
- Bromfield, E.B.; Dworetzky, B.A.; Wyszynski, D.F.; Smith, C.R.; Baldwin, E.J.; Holmes, L.B. Valproate teratogenicity and epilepsy syndrome. Epilepsia 2008, 49, 2122–2124. [Google Scholar] [CrossRef]
- Rasalam, A.D.; Hailey, H.; Williams, J.H.; Moore, S.J.; Turnpenny, P.D.; Lloyd, D.J.; Dean, J.C. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol. 2005, 47, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Binkerd, P.E.; Rowland, J.M.; Nau, H.; Hendrickx, A.G. Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague-Dawley rats. Fundam. Appl. Toxicol. 1988, 11, 485–493. [Google Scholar] [CrossRef]
- Wagner, G.C.; Reuhl, K.R.; Cheh, M.; McRae, P.; Halladay, A.K. A new neurobehavioral model of autism in mice: Pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord. 2006, 36, 779–793. [Google Scholar] [CrossRef]
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Nelson, S.; Romano, J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996, 370, 247–261. [Google Scholar] [CrossRef]
- Ingram, J.L.; Peckham, S.M.; Tisdale, B.; Rodier, P.M. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 2000, 22, 319–324. [Google Scholar] [CrossRef]
- Welsh, J.P.; Ahn, E.S.; Placantonakis, D.G. Is autism due to brain desynchronization? Int. J. Dev. Neurosci. 2005, 23, 253–263. [Google Scholar] [CrossRef]
- Weinstein-Fudim, L.; Ergaz, Z. Prevention or Amelioration of Autism-Like Symptoms in Animal Models: Will it Bring Us Closer to Treating Human ASD? Int. J. Mol. Sci. 2019, 20, 1074. [Google Scholar]
- Choi, C.S.; Gonzales, E.L.; Kim, K.C.; Yang, S.M.; Kim, J.W.; Mabunga, D.F.; Cheong, J.H.; Han, S.H.; Bahn, G.H.; Shin, C.Y. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci. Rep. 2016, 6, 36250. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [Green Version]
- Detich, N.; Bovenzi, V.; Szyf, M. Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 2003, 278, 27586–27592. [Google Scholar] [CrossRef] [Green Version]
- Wiltse, J. Mode of action: Inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of beta-catenin—Developmental effects of valproic acid. Crit. Rev. Toxicol. 2005, 35, 727–738. [Google Scholar] [CrossRef]
- Gurvich, N.; Berman, M.G.; Wittner, B.S.; Gentleman, R.C.; Klein, P.S.; Green, J.B. Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J. 2005, 19, 1166–1168. [Google Scholar] [CrossRef]
- Menegola, E.; Di Renzo, F.; Broccia, M.L.; Giavini, E. Inhibition of histone deacetylase as a new mechanism of teratogenesis. Birth Defects Res. Part C Embryo Today 2006, 78, 345–353. [Google Scholar] [CrossRef]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.S.; Wu, X.; Pang, H.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; Hong, J.S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Quina, A.S.; Buschbeck, M.; Di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nature 2005, 436, 1103–1106. [Google Scholar] [CrossRef]
- Shafique, S.; Winn, L.M. Gestational valproic acid exposure induces epigenetic modifications in murine decidua. Placenta 2021, 107, 31–40. [Google Scholar] [CrossRef]
- Felisbino, M.B.; Ziemann, M.; Khurana, I.; Okabe, J.; Al-Hasani, K.; Maxwell, S.; Harikrishnan, K.N.; de Oliveira, C.B.M.; Mello, M.L.S.; El-Osta, A. Valproic acid influences the expression of genes implicated with hyperglycaemia-induced complement and coagulation pathways. Sci. Rep. 2021, 11, 2163. [Google Scholar] [CrossRef]
- Roy, M.; Leclerc, D.; Wu, Q.; Gupta, S.; Kruger, W.D.; Rozen, R. Valproic acid increases expression of methylenetetrahydrofolate reductase (MTHFR) and induces lower teratogenicity in MTHFR deficiency. J. Cell. Biochem. 2008, 105, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Tsujimura, K.; Otsuka, I.M.; Irie, K.; Igarashi, K.; Nakashima, K.; Zhao, X. VPA alleviates neurological deficits and restores gene expression in a mouse model of Rett syndrome. PLoS ONE 2014, 9, e100215. [Google Scholar] [CrossRef] [Green Version]
- Cohen, O.S.; Varlinskaya, E.I.; Wilson, C.A.; Glatt, S.J.; Mooney, S.M. Acute prenatal exposure to a moderate dose of valproic acid increases social behavior and alters gene expression in rats. Int. J. Dev. Neurosci. 2013, 31, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Jacob, J.; Ribes, V.; Moore, S.; Constable, S.C.; Sasai, N.; Gerety, S.S.; Martin, D.J.; Sergeant, C.P.; Wilkinson, D.G.; Briscoe, J. Valproic acid silencing of ascl1b/Ascl1 results in the failure of serotonergic differentiation in a zebrafish model of fetal valproate syndrome. Dis. Models Mech. 2014, 7, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef]
- Jahani, M.; Khanahmad, H.; Nikpour, P. Evaluation of the Effects of Valproic Acid Treatment on Cell Survival and Epithelial-Mesenchymal Transition-Related Features of Human Gastric Cancer Cells. J. Gastrointest. Cancer 2021, 52, 676–681. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Gravemann, U.; Volland, J.; Nau, H. Hydroxamic acid and fluorinated derivatives of valproic acid: Anticonvulsant activity, neurotoxicity and teratogenicity. Neurotoxicol. Teratol. 2008, 30, 390–394. [Google Scholar] [CrossRef]
- Modi, H.R.; Basselin, M.; Rapoport, S.I. Valnoctamide, a non-teratogenic amide derivative of valproic acid, inhibits arachidonic acid activation in vitro by recombinant acyl-CoA synthetase-4. Bipolar Disord. 2014, 16, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Binder, T.; Salaj, P.; Zima, T.; Vítek, L. Randomized prospective comparative study of ursodeoxycholic acid and S-adenosyl-L-methionine in the treatment of intrahepatic cholestasis of pregnancy. J. Perinat. Med. 2006, 34, 383–391. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Panmanee, J.; Bradley-Clarke, J.; Mato, J.M.; O’Neill, P.M.; Antonyuk, S.V.; Hasnain, S.S. Control and regulation of S-Adenosylmethionine biosynthesis by the regulatory β subunit and quinolone-based compounds. FEBS J. 2019, 286, 2135–2154. [Google Scholar] [CrossRef]
- Cantoni, G.L. Biological methylation: Selected aspects. Annu. Rev. Biochem. 1975, 44, 435–451. [Google Scholar] [CrossRef]
- Mato, J.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- De Berardis, D.; Orsolini, L.; Serroni, N.; Girinelli, G.; Iasevoli, F.; Tomasetti, C.; de Bartolomeis, A.; Mazza, M.; Valchera, A.; Fornaro, M.; et al. A comprehensive review on the efficacy of S-Adenosyl-L-methionine in Major Depressive Disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 35–44. [Google Scholar] [CrossRef]
- Cuomo, A.; Beccarini Crescenzi, B.; Bolognesi, S.; Goracci, A.; Koukouna, D.; Rossi, R.; Fagiolini, A. S-Adenosylmethionine (SAMe) in major depressive disorder (MDD): A clinician-oriented systematic review. Ann. Gen. Psychiatry 2020, 19, 50. [Google Scholar] [CrossRef]
- Sarris, J.; Murphy, J.; Stough, C.; Mischoulon, D.; Bousman, C.; MacDonald, P.; Adams, L.; Nazareth, S.; Oliver, G.; Cribb, L.; et al. S-Adenosylmethionine (SAMe) monotherapy for depression: An 8-week double-blind, randomised, controlled trial. Psychopharmacology 2020, 237, 209–218. [Google Scholar] [CrossRef]
- Jacobsen, S.; Danneskiold-samsøe, B.; Andersen, R.B. Oral S-adenosylmethionine in Primary Fibromyalgia. Double-blind Clinical Evaluation. Scand. J. Rheumatol. 1991, 20, 294–302. [Google Scholar] [CrossRef]
- Shekim, W.O.; Antun, F.; Hanna, G.L.; McCracken, J.T.; Hess, E.B. S-adenosyl-L-methionine (SAM) in adults with ADHD, RS: Preliminary results from an open trial. Psychopharmacol. Bull. 1990, 26, 249–253. [Google Scholar]
- Di Rocco, A.; Rogers, J.D.; Brown, R.; Werner, P.; Bottiglieri, T. S-Adenosyl-Methionine improves depression in patients with Parkinson’s disease in an open-label clinical trial. Mov. Disord. 2000, 15, 1225–1229. [Google Scholar] [CrossRef]
- Beauchamp, L.C.; Liu, X.M.; Sedjahtera, A.; Bogeski, M.; Vella, L.J.; Bush, A.I.; Adlard, P.A.; Barnham, K.J. S-Adenosylmethionine Rescues Cognitive Deficits in the rTg4510 Animal Model by Stabilizing Protein Phosphatase 2A and Reducing Phosphorylated Tau. J. Alzheimer’s Dis. JAD 2020, 77, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- König, B. A long-term (two years) clinical trial with S-adenosylmethionine for the treatment of osteoarthritis. Am. J. Med. 1987, 83, 89–94. [Google Scholar] [CrossRef]
- Friedel, H.A.; Goa, K.L.; Benfield, P. S-Adenosyl-L-Methionine. Drugs 1989, 38, 389–416. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Pérez, V.; Muñoz, M.; Corpa, J.M.; Abad, M.; Carbajo, M.T. Effects of S-adenosylmethionine on hepatic regeneration after partial hepatectomy in the rat. J. Physiol. Biochem. 2003, 59, 63–64. [Google Scholar] [CrossRef]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.-Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Zhao, C. Therapeutic effect of adenosylmethionine on viral hepatitis and related factors inducing diseas. Am. J. Transl. Res. 2021, 13, 9485–9494. [Google Scholar]
- Ilisso, C.P.; Sapio, L.; Delle Cave, D.; Illiano, M.; Spina, A.; Cacciapuoti, G.; Naviglio, S.; Porcelli, M. S-Adenosylmethionine Affects ERK1/2 and Stat3 Pathways and Induces Apotosis in Osteosarcoma Cells. J. Cell. Physiol. 2016, 231, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pépin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2018, 9, 5169–5183. [Google Scholar] [CrossRef] [Green Version]
- Ilisso, C.P.; Castellano, M.; Zappavigna, S.; Lombardi, A.; Vitale, G.; Dicitore, A.; Cacciapuoti, G.; Caraglia, M.; Porcelli, M. The methyl donor S-adenosylmethionine potentiates doxorubicin effects on apoptosis of hormone-dependent breast cancer cell lines. Endocrine 2015, 50, 212–222. [Google Scholar] [CrossRef]
- Yang, J.; He, Y.; Du, Y.X.; Tang, L.L.; Wang, G.J.; Fawcett, J.P. Pharmacokinetic properties of S-adenosylmethionine after oral and intravenous administration of its tosylate disulfate salt: A multiple-dose, open-label, parallel-group study in healthy Chinese volunteers. Clin. Ther. 2009, 31, 311–320. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.J.; Ryckman, K.K. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab. Syndr. Obes. 2015, 8, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, H.; Sanchez-Cabanillas, C.; Cebola, I. Epigenetics of Hepatic Insulin Resistance. Front. Endocrinol. 2021, 12, 504. [Google Scholar] [CrossRef] [PubMed]
- Frías-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef]
- Corso-Díaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef]
- Dindot, S.V.; Person, R.; Strivens, M.; Garcia, R.; Beaudet, A.L. Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions. Genome Res. 2009, 19, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Matouk, C.C.; Marsden, P.A. Epigenetic regulation of vascular endothelial gene expression. Circ. Res. 2008, 102, 873–887. [Google Scholar] [CrossRef]
- Golding, M.C.; Williamson, G.L.; Stroud, T.K.; Westhusin, M.E.; Long, C.R. Examination of DNA methyltransferase expression in cloned embryos reveals an essential role for Dnmt1 in bovine development. Mol. Reprod. Dev. 2011, 78, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Warner, M.J.; Ozanne, S.E. Mechanisms involved in the developmental programming of adulthood disease. Biochem. J. 2010, 427, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, S.; Ishii, K.; Tajima, A.; Iga, J.; Kinoshita, M.; Watanabe, S.; Umehara, H.; Fuchikami, M.; Okada, S.; Boku, S.; et al. Blood diagnostic biomarkers for major depressive disorder using multiplex DNA methylation profiles: Discovery and validation. Epigenetics 2015, 10, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indika, N.-L.R.; Deutz, N.E.P.; Engelen, M.P.K.J.; Peiris, H.; Wijetunge, S.; Perera, R. Sulfur amino acid metabolism and related metabotypes of autism spectrum disorder: A review of biochemical evidence for a hypothesis. Biochimie 2021, 184, 143–157. [Google Scholar] [CrossRef]
- Varela-Rey, M.; Iruarrizaga-Lejarreta, M.; Lozano, J.J.; Aransay, A.M.; Fernandez, A.F.; Lavin, J.L.; Mósen-Ansorena, D.; Berdasco, M.; Turmaine, M.; Luka, Z.; et al. S-adenosylmethionine Levels Regulate the Schwann Cell DNA Methylome. Neuron 2014, 81, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.J.; Shivapurkar, N.; Poirier, L.A. Hypomethylation of hepatic nuclear DNA in rats fed with a carcinogenic methyl-deficient diet. Biochem. J. 1984, 218, 987–990. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Meletis, K.; Fu, D.; Jhaveri, S.; Jaenisch, R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet Enzymes Compromises Proper Differentiation of Embryonic Stem Cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slieker, R.C.; Roost, M.S.; van Iperen, L.; Suchiman, H.E.D.; Tobi, E.W.; Carlotti, F.; de Koning, E.J.P.; Slagboom, P.E.; Heijmans, B.T.; Chuva de Sousa Lopes, S.M. DNA Methylation Landscapes of Human Fetal Development. PLoS Genet. 2015, 11, e1005583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Zheng, Y.L.; Xu, C.Q.; Gu, L.Z.; Ding, Z.L.; Qin, L.; Wang, Y.; Fu, R.; Wan, Y.F.; Hu, C.P. Valproic acid (VPA) enhances cisplatin sensitivity of non-small cell lung cancer cells via HDAC2 mediated down regulation of ABCA1. Biol. Chem. 2017, 398, 785–792. [Google Scholar] [CrossRef]
- Peñagaricano, F.; Souza, A.H.; Carvalho, P.D.; Driver, A.M.; Gambra, R.; Kropp, J.; Hackbart, K.S.; Luchini, D.; Shaver, R.D.; Wiltbank, M.C.; et al. Effect of maternal methionine supplementation on the transcriptome of bovine preimplantation embryos. PLoS ONE 2013, 8, e72302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei Saadi, H.A.; Gagné, D.; Fournier, É.; Baldoceda Baldeon, L.M.; Sirard, M.A.; Robert, C. Responses of bovine early embryos to S-adenosyl methionine supplementation in culture. Epigenomics 2016, 8, 1039–1060. [Google Scholar] [CrossRef]
- Cooney, C.A.; Dave, A.A.; Wolff, G.L. Maternal Methyl Supplements in Mice Affect Epigenetic Variation and DNA Methylation of Offspring. J. Nutr. 2002, 132, 2393S–2400S. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Jan Veenstra, G.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66 (Suppl. 1), S7–S11. [Google Scholar] [CrossRef] [Green Version]
- Duhl, D.M.; Vrieling, H.; Miller, K.A.; Wolff, G.L.; Barsh, G.S. Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 1994, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Weinstein-Fudim, L.; Ergaz, Z.; Szyf, M.; Ornoy, A. Prenatal S-Adenosine Methionine (SAMe) Induces Changes in Gene Expression in the Brain of Newborn Mice That Are Prevented by Co-Administration of Valproic Acid (VPA). Int. J. Mol. Sci. 2020, 21, 2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornoy, A.; Becker, M.; Weinstein-Fudim, L.; Ergaz, Z. S-Adenosine Methionine (SAMe) and Valproic Acid (VPA) as Epigenetic Modulators: Special Emphasis on their Interactions Affecting Nervous Tissue during Pregnancy. Int. J. Mol. Sci. 2020, 21, 3721. [Google Scholar] [CrossRef] [PubMed]
- Feder, Y.; Nesher, E.; Ogran, A.; Kreinin, A.; Malatynska, E.; Yadid, G.; Pinhasov, A. Selective breeding for dominant and submissive behavior in Sabra mice. J. Affect. Disord. 2010, 126, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Pinhasov, A.; Ornoy, A. Animal Models of Depression: What Can They Teach Us about the Human Disease? Diagnostics 2021, 11, 123. [Google Scholar] [CrossRef]
- Becker, M.; Abaev, K.; Pinhasov, A.; Ornoy, A. S-adenosyl methionine alleviates depression like behavior in a mouse model of social hierarchy. J. Behav. Brain Res. 2022; article under review. [Google Scholar]
- Frezza, M.; Centini, G.; Cammareri, G.; Le Grazie, C.; Di Padova, C. S-adenosylmethionine for the treatment of intrahepatic cholestasis of pregnancy. Results of a controlled clinical trial. Hepato-Gastroenterol. 1990, 37 (Suppl. 2), 122–125. [Google Scholar]
- Coltorti, M.; Bortolini, M.; Di Padova, C. A review of the studies on the clinical use of S-adenosylmethionine (SAMe) for the symptomatic treatment of intrahepatic cholestasis. Methods Find. Exp. Clin. Pharmacol. 1990, 12, 69–78. [Google Scholar]
- Hardy, M.L.; Coulter, I.; Morton, S.C.; Favreau, J.; Venuturupalli, S.; Chiappelli, F.; Rossi, F.; Orshansky, G.; Jungvig, L.K.; Roth, E.A.; et al. S-adenosyl-L-methionine for treatment of depression, osteoarthritis, and liver disease. Evid. Rep./Technol. Assess. 2003, 64, 1–3. [Google Scholar]
- Cozens, D.D.; Barton, S.J.; Clark, R.; Gibson, W.A.; Hughes, E.W.; Masters, R.E.; Offer, J.M.; Perkin, C.J.; Stramentinoli, G. Reproductive toxicity studies of ademetionine. Arzneim. -Forsch. 1988, 38, 1625–1629. [Google Scholar]
- Zeisel, S.H.; Blusztajn, J.K. Choline and human nutrition. Annu. Rev. Nutr. 1994, 14, 269–296. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Klatt, K.C.; Caudill, M.A. Choline. Adv. Nutr. 2018, 9, 58–60. [Google Scholar] [CrossRef] [Green Version]
- Caudill, M.A. Pre- and postnatal health: Evidence of increased choline needs. J. Am. Diet. Assoc. 2010, 110, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.L.; Wurtman, R.J. Brain acetylcholine: Control by dietary choline. Science 1976, 191, 561–562. [Google Scholar] [CrossRef]
- Abreu-Villaca, Y.; Filgueiras, C.C.; Manhaes, A.C. Developmental aspects of the cholinergic system. Behav. Brain Res. 2011, 221, 367–378. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Niculescu, M.D. Perinatal choline influences brain structure and function. Nutr. Rev. 2006, 64, 197–203. [Google Scholar] [CrossRef]
- Hoffmann, L.; Brauers, G.; Gehrmann, T.; Häussinger, D.; Mayatepek, E.; Schliess, F.; Schwahn, B.C. Osmotic regulation of hepatic betaine metabolism. Am. J. Physiology. Gastrointest. Liver Physiol. 2013, 304, G835–G846. [Google Scholar] [CrossRef] [Green Version]
- Traiffort, E.; O’Regan, S.; Ruat, M. The choline transporter-like family SLC44: Properties and roles in human diseases. Mol. Asp. Med. 2013, 34, 646–654. [Google Scholar] [CrossRef]
- Lu, S.C.; Alvarez, L.; Huang, Z.-Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albright, C.D.; Tsai, A.Y.; Friedrich, C.B.; Mar, M.H.; Zeisel, S.H. Choline availability alters embryonic development of the hippocampus and septum in the rat. Brain Res. Dev. Brain Res. 1999, 113, 13–20. [Google Scholar] [CrossRef]
- Albright, C.D.; Mar, M.H.; Friedrich, C.B.; Brown, E.C.; Zeisel, S.H. Maternal choline availability alters the localization of p15Ink4B and p27Kip1 cyclin-dependent kinase inhibitors in the developing fetal rat brain hippocampus. Dev. Neurosci. 2001, 23, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006, 20, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, S.T.C.; King, J.H.; Grenier, J.K.; Yan, J.; Jiang, X.; Roberson, M.S.; Caudill, M.A. Maternal Choline Supplementation during Normal Murine Pregnancy Alters the Placental Epigenome: Results of an Exploratory Study. Nutrients 2018, 10, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, S.T.C.; King, J.H.; Yan, J.; Jiang, X.; Wei, E.; Fomin, V.G.; Roberson, M.S.; Caudill, M.A. Maternal choline supplementation during murine pregnancy modulates placental markers of inflammation, apoptosis and vascularization in a fetal sex-dependent manner. Placenta 2017, 53, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.; MacDonald, C.J.; Williams, C.L.; Meck, W.H. Prenatal choline supplementation alters the timing, emotion, and memory performance (TEMP) of adult male and female rats as indexed by differential reinforcement of low-rate schedule behavior. Learn Mem. 2008, 15, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Meck, W.H.; Williams, C.L. Choline supplementation during prenatal development reduces proactive interference in spatial memory. Brain Res. Dev. Brain Res. 1999, 118, 51–59. [Google Scholar] [CrossRef]
- Trujillo-Gonzalez, I.; Friday, W.B.; Munson, C.A.; Bachleda, A.; Weiss, E.R.; Alam, N.M.; Sha, W.; Zeisel, S.H.; Surzenko, N. Low availability of choline in utero disrupts development and function of the retina. FASEB J. 2019, 33, 9194–9209. [Google Scholar] [CrossRef]
- May, P.A.; Chambers, C.D.; Kalberg, W.O.; Zellner, J.; Feldman, H.; Buckley, D.; Kopald, D.; Hasken, J.M.; Xu, R.; Honerkamp-Smith, G.; et al. Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA 2018, 319, 474–482. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Riley, E.P.; Charness, M.E. Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorder. Lancet Neurol. 2019, 18, 760–770. [Google Scholar] [CrossRef]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.P.; Cynader, M.S.; et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin 2016, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Steane, S.E.; Fielding, A.M.; Kent, N.L.; Andersen, I.; Browne, D.J.; Tejo, E.N.; Gardebjer, E.M.; Kalisch-Smith, J.I.; Sullivan, M.A.; Moritz, K.M.; et al. Maternal choline supplementation in a rat model of periconceptional alcohol exposure: Impacts on the fetus and placenta. Alcohol Clin. Exp. Res. 2021, 45, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Sawant, O.B.; Birch, S.M.; Goodlett, C.R.; Cudd, T.A.; Washburn, S.E. Maternal choline supplementation mitigates alcohol-induced fetal cranio-facial abnormalities detected using an ultrasonographic examination in a sheep model. Alcohol 2019, 81, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.W.; Carter, R.C.; Molteno, C.D.; Stanton, M.E.; Herbert, J.S.; Lindinger, N.M.; Lewis, C.E.; Dodge, N.C.; Hoyme, H.E.; Zeisel, S.H.; et al. Efficacy of Maternal Choline Supplementation During Pregnancy in Mitigating Adverse Effects of Prenatal Alcohol Exposure on Growth and Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol Clin. Exp. Res. 2018, 42, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Warton, F.L.; Molteno, C.D.; Warton, C.M.R.; Wintermark, P.; Lindinger, N.M.; Dodge, N.C.; Zollei, L.; van der Kouwe, A.J.W.; Carter, R.C.; Jacobson, J.L.; et al. Maternal choline supplementation mitigates alcohol exposure effects on neonatal brain volumes. Alcohol Clin. Exp. Res. 2021, 45, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Kable, J.A.; Coles, C.D.; Keen, C.L.; Uriu-Adams, J.Y.; Jones, K.L.; Yevtushok, L.; Kulikovsky, Y.; Wertelecki, W.; Pedersen, T.L.; Chambers, C.D.; et al. The impact of micronutrient supplementation in alcohol-exposed pregnancies on information processing skills in Ukrainian infants. Alcohol 2015, 49, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.G.; Hunter, S.K.; McCarthy, L.; Beuler, J.; Hutchison, A.K.; Wagner, B.D.; Leonard, S.; Stevens, K.E.; Freedman, R. Perinatal choline effects on neonatal pathophysiology related to later schizophrenia risk. Am. J. Psychiatry 2013, 170, 290–298. [Google Scholar] [CrossRef]
- Boeke, C.E.; Gillman, M.W.; Hughes, M.D.; Rifas-Shiman, S.L.; Villamor, E.; Oken, E. Choline intake during pregnancy and child cognition at age 7 years. Am. J. Epidemiol. 2013, 177, 1338–1347. [Google Scholar] [CrossRef] [Green Version]
- Caudill, M.A.; Strupp, B.J.; Muscalu, L.; Nevins, J.E.H.; Canfield, R.L. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB J. 2018, 32, 2172–2180. [Google Scholar] [CrossRef] [Green Version]
- Freedman, R.; Hunter, S.K.; Law, A.J.; Wagner, B.D.; D’Alessandro, A.; Christians, U.; Noonan, K.; Wyrwa, A.; Hoffman, M.C. Higher Gestational Choline Levels in Maternal Infection Are Protective for Infant Brain Development. J. Pediatrics 2019, 208, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, C.L.; Goldman, B.D.; Fischer, L.M.; da Costa, K.A.; Reznick, J.S.; Zeisel, S.H. Phosphatidylcholine supplementation in pregnant women consuming moderate-choline diets does not enhance infant cognitive function: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2012, 96, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.R.; Fuglestad, A.J.; Eckerle, J.K.; Fink, B.A.; Hoecker, H.L.; Boys, C.J.; Radke, J.P.; Kroupina, M.G.; Miller, N.C.; Brearley, A.M.; et al. Choline supplementation in children with fetal alcohol spectrum disorders: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2015, 102, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.K.; Gangisetty, O.; Wozniak, J.R.; Eckerle, J.K.; Georgieff, M.K.; Foroud, T.M.; Wetherill, L.; Wertelecki, W.; Chambers, C.D.; Riley, E.; et al. Persistent Changes in Stress-Regulatory Genes in Pregnant Women or Children Exposed Prenatally to Alcohol. Alcohol Clin. Exp. Res. 2019, 43, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Risbud, R.D.; Mattson, S.N.; Chambers, C.D.; Thomas, J.D. Randomized, double-blind, placebo-controlled clinical trial of choline supplementation in school-aged children with fetal alcohol spectrum disorders. Am. J. Clin. Nutr. 2016, 104, 1683–1692. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| First Editor | Animals | VPA Treatment | Outcomes |
|---|---|---|---|
| Roy et al., 2008 | Mice with reduced activity of Mthfr gene (Mthfr+/−) | Single dose of 300 mg/kg VPA on day 8.5 of pregnancy | Increase in the expression of Mthfr gene and normalization of MTHFR protein; improved fetal outcome compared to treated normal mice |
| Guo et al., 2014 | Six-week-old mice with MECP2 deficiency (Rett syndrome) | Daily injections of 350 mg/kg VPA for 2 weeks | Enhanced expression of MeCP2 gene and increased MeCP2 protein with improvement in the clinical symptoms of Rett syndrome |
| Cohen et al., 2013 | Normal rats | 350 mg/kg VPA on day 13 of gestation in normal rats | Increased expression in the brain of genes encoding for acetylation-sensitive proteins and increased social investigation and play fighting |
| Jacob et al., 2014 | Zebrafish | 0.625 mM VPA in water | Downregulation of the proneuronal gene ascl1b and inhibition of serotonin synthesis in the brain |
| Ornoy et al., 2019 | 4-day-old ICR mice | 300 mg/kg VPA injected to 4-day-old offspring or VPA and SAMe 30 mg/kg during postnatal days 4–6 | Induction of autistic-like behavior and increased oxidative stress in the prefrontal cortex ameliorated by SAMe |
| Weinstein et al., 2019 | 4-day-old ICR mice | 300 mg/kg VPA injected to 4-day-old offspring or VPA and SAMe 30 mg/kg during postnatal days 4–6 | VPA induced changes in the expression of neurophysiologic and neuropathlogic genes in the prefrontal cortex that were reversed to normal by SAMe |
| First Editor | Animal/Human | Treatment | Outcomes |
|---|---|---|---|
| Peñagaricano et al., 2013 | Holstein cows early embryo | Methionine supplementation during follicular phase and early embryo development, until day 7. | In total, 276 of the 10,662 genes analyzed showed significant differences following treatment. Maternal methionine supplementation resulted in decreased expression of most genes, reduced transcription, and increased methylation of specific genes. |
| Shojaei Saadi et al., 2002 | In vitro cultured bovine embryos | SAMe treatment from the eight-cell stage to the blastocyst stage. | SAMe induced genome-wide hypermethylation mainly in exonic regions and in CpG islands. Differentially expressed genes were associated with the response to nutrients and developmental processes. |
| Cooney et al., 2002 | Viable yellow agouti (A(vy)) mouse strains that carry mutant alleles at the agouti locus | Methyl donor diets: betaine, choline, folic acid, B12 with or without methionine, and zinc during gestation. | The increase in offspring DNA methylation state resulted in a change from a yellow coat color distribution to a pseudo agouti coat as a result of alterations in the gene expression. |
| Weinstein et al., 2020 | ICR mice | Oral SAMe given to DAMs on days 12–15 of gestation. | Significant gender-related changes in the expression of many genes in the brain of 1-day-old offspring. The most prominent changes in gene expression were Vegfa and its receptor Flt1. |
| Becker et al., 2021 | Sub mice derived from Sabra | Oral gavage with SAMe (20 mg/kg) on gestational days 12–14. | Improved depression-like behavior at adulthood, especially in the three chamber test for sociability. Increased expression of Vegfa and its receptor Flt1 genes in the prefrontal cortex at 90 days of age. |
| Tomáš Binder et al., 1990 | Pregnant Women with intrahepatic cholestasis of pregnancy (ICP) | SAMe for the treatment of intrahepatic cholestasis. | No adverse effects were noted on the fetuses or neonates. |
| Frezza, M et al., 1990 | Thirty patients in the last trimester of pregnancy with intrahepatic cholestasis of pregnancy (ICP) | SAMe (800 mg/day i.v.) or placebo until delivery for a mean period of 18 days. | No adverse reactions on mother or child were recorded during SAMe treatment, and at follow-up of the children. Possibly decreased rate of prematurity compared to placebo treated. |
| Coltorti et al., 1990 | Eighteen patients with intrahepatic cholestasis of pregnancy (ICP) | SAMe 900 mg/day or placebo was administered by daily intravenous infusions for 20 days. | No relevant adverse reactions were detected. All newborns had Apgar scores greater than 7 and normal postnatal development. |
| First Editor | Rodents | Treatment | Outcome |
|---|---|---|---|
| Albright et al., 1999 | Sprague-Dawley rats fetal brain sections were collected on days 18 and 20 of pregnancy | Varying levels of dietary choline for 6 days from gestational day 12. | Choline deficiency-related brain changes including an increased number of apoptotic cells in the dentate gyrus and increased expression of TOAD-64, a neuronal differentiation marker, in the hippocampus. |
| Albright et al., 2001 | Sprague-Dawley rats fetal brain sections were collected on days 18 and 20 of pregnancy | Varying levels of dietary choline for 6 days from gestational day 12. | Maternal dietary choline deficiency changed the localization of p15Ink4B and p27Kip1 cyclin-dependent kinase inhibitors in the offspring hippocampus. |
| Mellott et al., 2004 | Sprague-Dawley rats | Pregnant rats fed a choline-supplemented diet for 8 days (between embryonic days 11 and 18) | Choline-supplemented rats showed evidence of a precocious capacity for the spatial navigation water maze task. Choline also in creased activation of mitogen-activated protein kinase (MAPK) and cAMP-response element binding protein (CREB) in the hippocampus. |
| Niculescu et al., 2006 | C57 BL/6 mice | Pregnant dams fed deficient or normal in choline content diet from days 12 to 17 of pregnancy. Fetal brains collected on embryonic day 17. | Choline deficiency increased protein levels of kinase-associated phosphatase (Kap) and p15INK4b (two cell cycle inhibitors) and decreased gene-specific DNA methylation in the offspring brain. |
| Cheng et al., 2008 | Sprague-Dawley rats | Pregnant rats fed normal choline (1.1 g/kg) or supplemental choline (5.0 g/kg) during embryonic days 12–17. Male and female offspring conducted behavioral training at 7 months of age. | Prenatal choline supplement-action was associated with an improvement of cognitive function, spatial memory, and attentional function |
| Kwan et al., 2018 | Swiss Albino mice | Pregnant mice fed a 1X (1.4 g choline chloride/kg diet) or 4X choline (5.6 g choline chloride/kg diet) diet from embryonic day 0.5. placentas collected on embryonic day 15.5. | High choline levels during gestation altered the expression of several imprinted genes in a sex-specific manner. |
| Trujillo-Gonzalez et al., 2019 | NestinCFPnuc Nestin-CreERT2, Ai9 and C57BL/6J mice | Pregnant dams randomly assigned to either adequate (1.4 g/kg of choline chloride) or low-choline diet (1.2 g/kg of choline chloride) administered starting at day 11.5 of pregnancy. | Low-choline diet during gestation was associated with disrupted retina development and visual function. |
| Steane et al., 2021 | Sprague Dawley rats | Female rats exposed to 12.5% ethanol from 4 days prior to 4 days after conception. From day 5 of pregnancy, dams were placed on different choline levels chow (1.6–7.2 g choline/kg chow). Fetuses and placentas were collected on day 20 of pregnancy for analysis. | Choline supplementation resulted in increased fetal weight by late gestation, ameliorating the deficits caused by maternal ethanol consumption |
| First Editor | Participants | Treatment | Outcomes |
|---|---|---|---|
| Ross et al., 2013 | 100 healthy pregnant women and their infants. | Phospohadtidycholine (approximately 900 mg choline/day) from the second trimester of pregnancy and 100 mg until the age of 3 months. | Infants treated with choline are significantly more likely to have normal cerebral inhibition at 5 weeks of age. |
| Kable et al., 2015 | Pregnant women with moderate/heavy alcohol use. | 750 mg choline (n = 37) multivitamin/mineral supplement (n = 23); combination of both treatments (n = 19), standard care (n = 35) | Multivitamin/mineral combined with choline supplementation significantly improved basic attentional regulation systems. |
| Jacobson et al., 2018 | 69 pregnant heavy drinker mothers and their infants. | 2000 mg choline (n = 34 or placebo (n = 35) from mid-pregnancy until delivery | Choline treatment improved weight, postnatal growth, cognition, and eye-blink conditioning at 6.5 and 12 months of age. |
| Caudil et al., 2018 | 26 pregnant women entering their third trimester until delivery, and their infants | Either 480 mg choline/d (n = 13) or 930 mg choline/d (n = 13). | Higher choline levels associated with faster information processing speed in infants at 4–13 months. |
| Freedman et al., 2019 | 162 Pregnant women with different infections at 16 week of gestation and their infants at 1 year of age | Maternal serum choline and betaine levels were measured at 16 week of gestation. | Higher gestational choline concentrations were associated with improved development of cerebral inhibition and cerebral regulation at the age of one year. |
| Warton et al., 2021 | 52 mothers with heavy drinking and their infants. | 2000 mg choline (n = 28) or placebo (n = 24) from mid-pregnancy until delivery. | Choline supplementation during pregnancy mitigated regional volume reductions in alcohol-exposed infants, with larger volumes associated with improved 12-month recognition memory. |
| Hunter et al., 2021 | 122 pregnant mothers and their children from 3 months of age; 48 children completed the assessment at 4 years of age. | Maternal serum choline and betaine levels were measured at 16 and 28 weeks of gestation. | Prenatal maternal choline levels were positively associated with higher processing speed and decreased problems in social withdrawal. |
| Cheatham et al., 2012 | 140 pregnant women assigned from 18 weeks of gestation, and their infants | 750 mg phosphatidyl-choline (n = 49) or placebo (n = 50) daily from 18 weeks of gestation until 90 days post-partum and their infants | No significant effect of phosphatidylcholine supplement detected on short-term visuospatial memory, long-term episodic memory, language development and global development at 10 and 12 months of age. |
| First Editor | Participants | Treatment | Outcomes |
|---|---|---|---|
| Wozniak et al., 2015 | 60 children aged 2.5–5 years at enrollment, with FASDs | 500 mg choline or placebo daily for 9 months | Choline significantly improved delayed sequential memory in 2–3-year-olds. |
| Sarkar et al., 2019 | 60 children aged 2.5–5 years at enrollment, with FASDs | 500 mg choline or a placebo daily for 9 months | Choline supplementation reduced DNA methylation of hPER2 and hPOMC genes and increased the expression of stress regulatory genes. |
| Wozniak et al., 2020 | Follow up of 31 children with FASDs mean age 8.6 years | 500 mg choline (n = 15) or a placebo (n = 16) daily for 9 months between 2.5 and 5 years | Choline significantly improved nonverbal intelligence, higher visual-spatial skills, working memory ability, and verbal memory, and decreased behavioral symptoms of attention deficit hyperactivity disorder. |
| Nguyen et al., 2016 | 55 children aged 5–10 years, with confirmed histories of heavy prenatal alcohol exposure. | 625 mg choline (n = 29) or placebo (n = 26) daily for 6 weeks | Choline supplementation did not improve cognitive performance in any domain. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ornoy, A.; Weinstein-Fudim, L.; Becker, M. SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies. Pharmaceuticals 2022, 15, 192. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020192
Ornoy A, Weinstein-Fudim L, Becker M. SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies. Pharmaceuticals. 2022; 15(2):192. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020192
Chicago/Turabian StyleOrnoy, Asher, Liza Weinstein-Fudim, and Maria Becker. 2022. "SAMe, Choline, and Valproic Acid as Possible Epigenetic Drugs: Their Effects in Pregnancy with a Special Emphasis on Animal Studies" Pharmaceuticals 15, no. 2: 192. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15020192