
Supercritical Fluid Chromatography—Tandem Mass Spectrometry for Rapid Quantification of Pentacyclic Triterpenoids in Plant Extracts

 , , ,
, , ,
Abstract
:1. Introduction
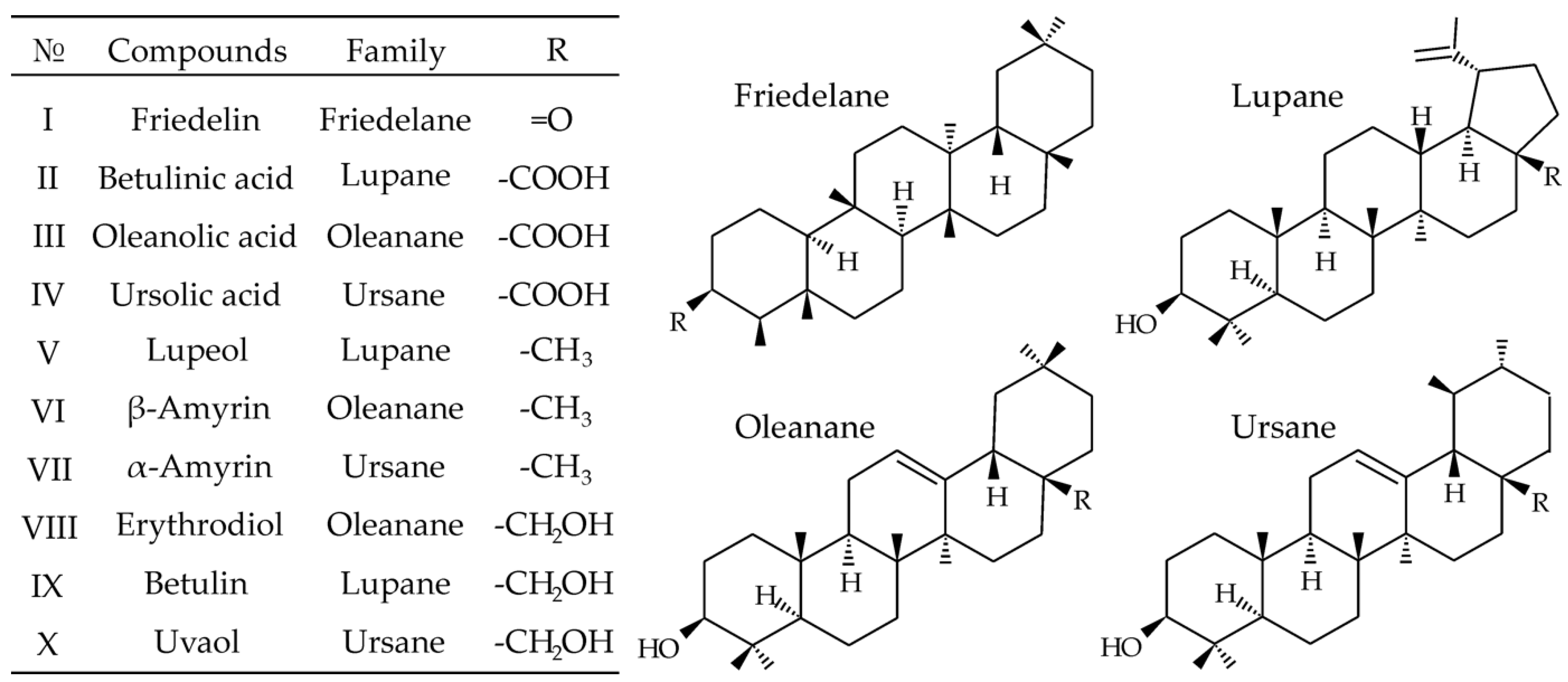
2. Results and Discussion
2.1. Tandem Mass Spectrometry
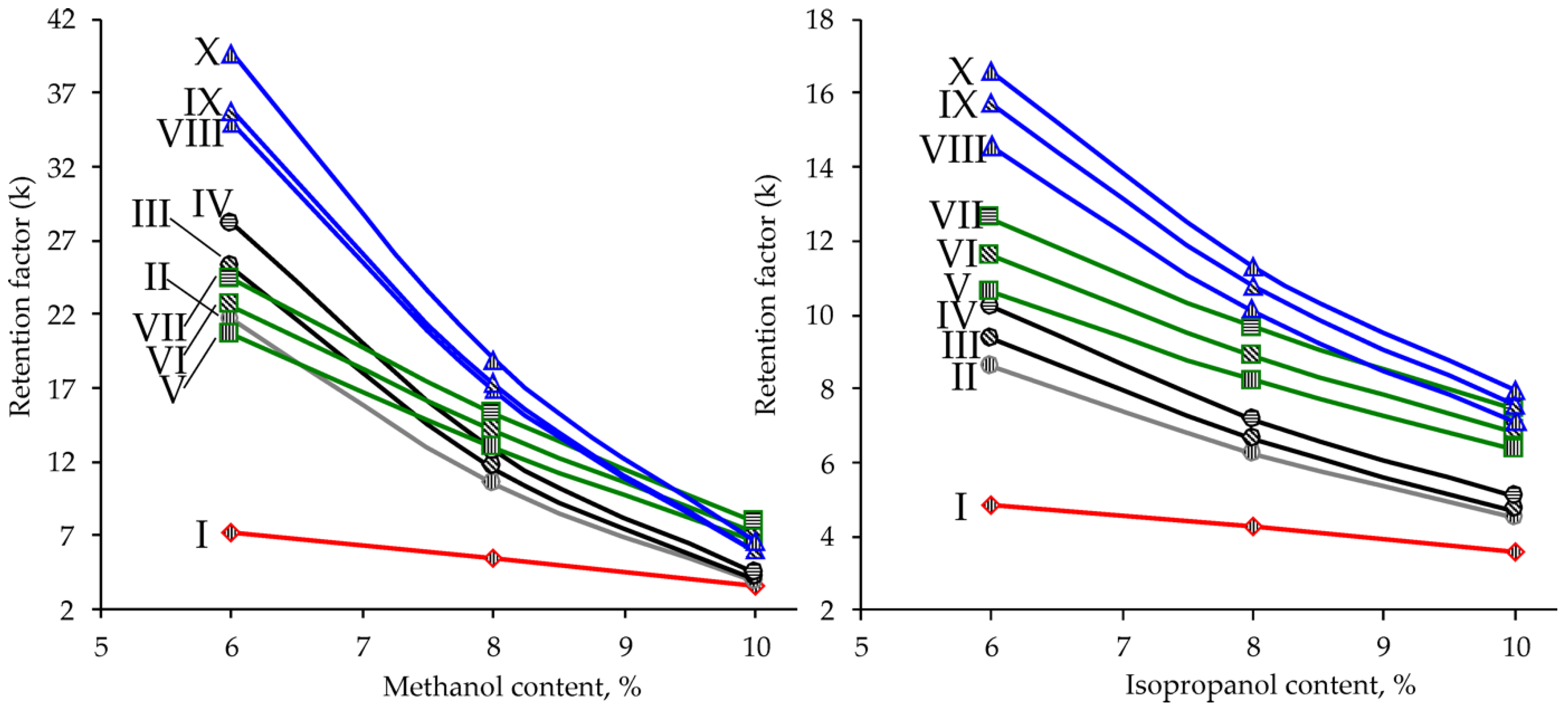
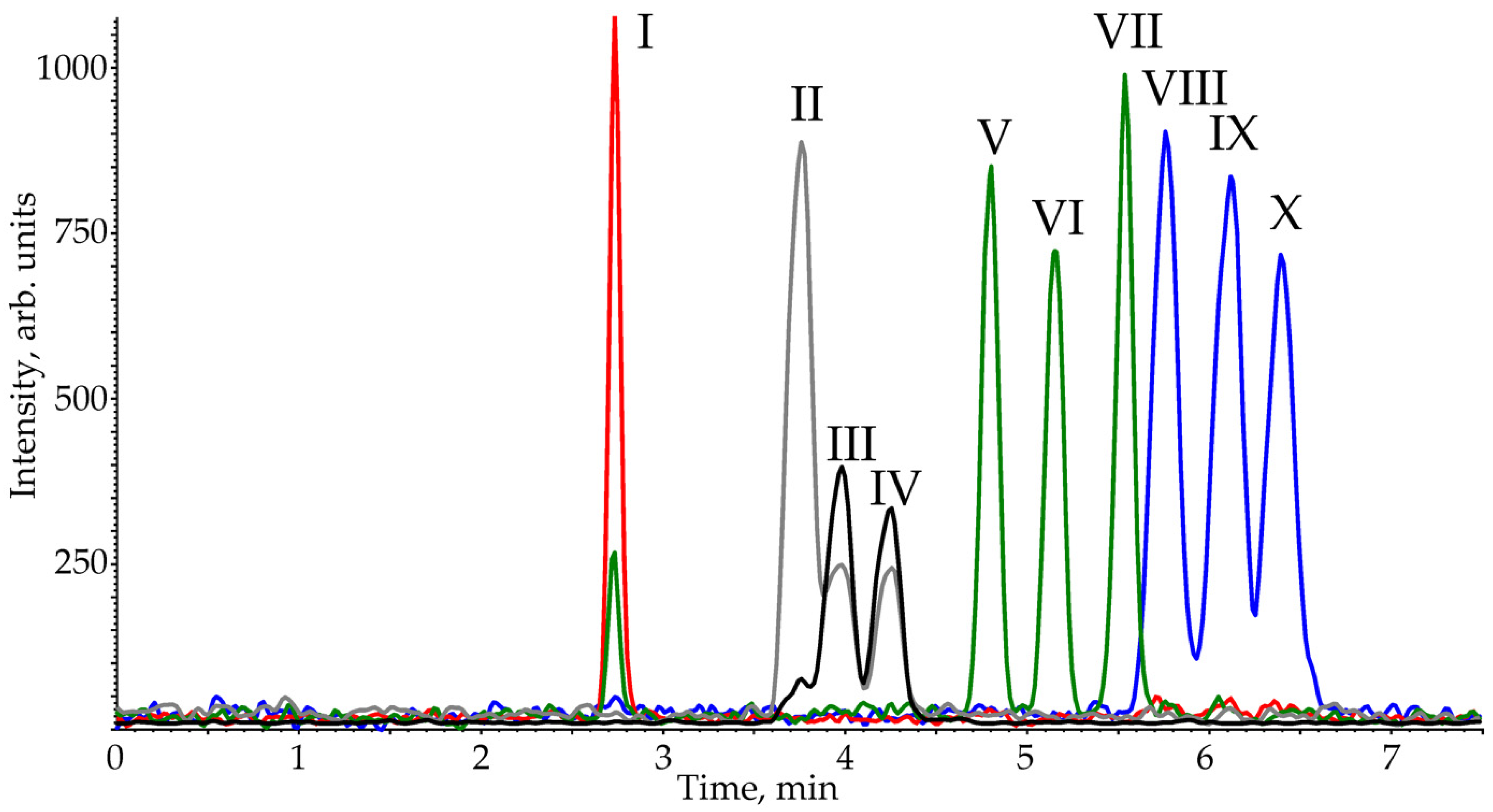
2.2. Chromatographic Separation and Column Screening
2.3. Quantification and Method Validation
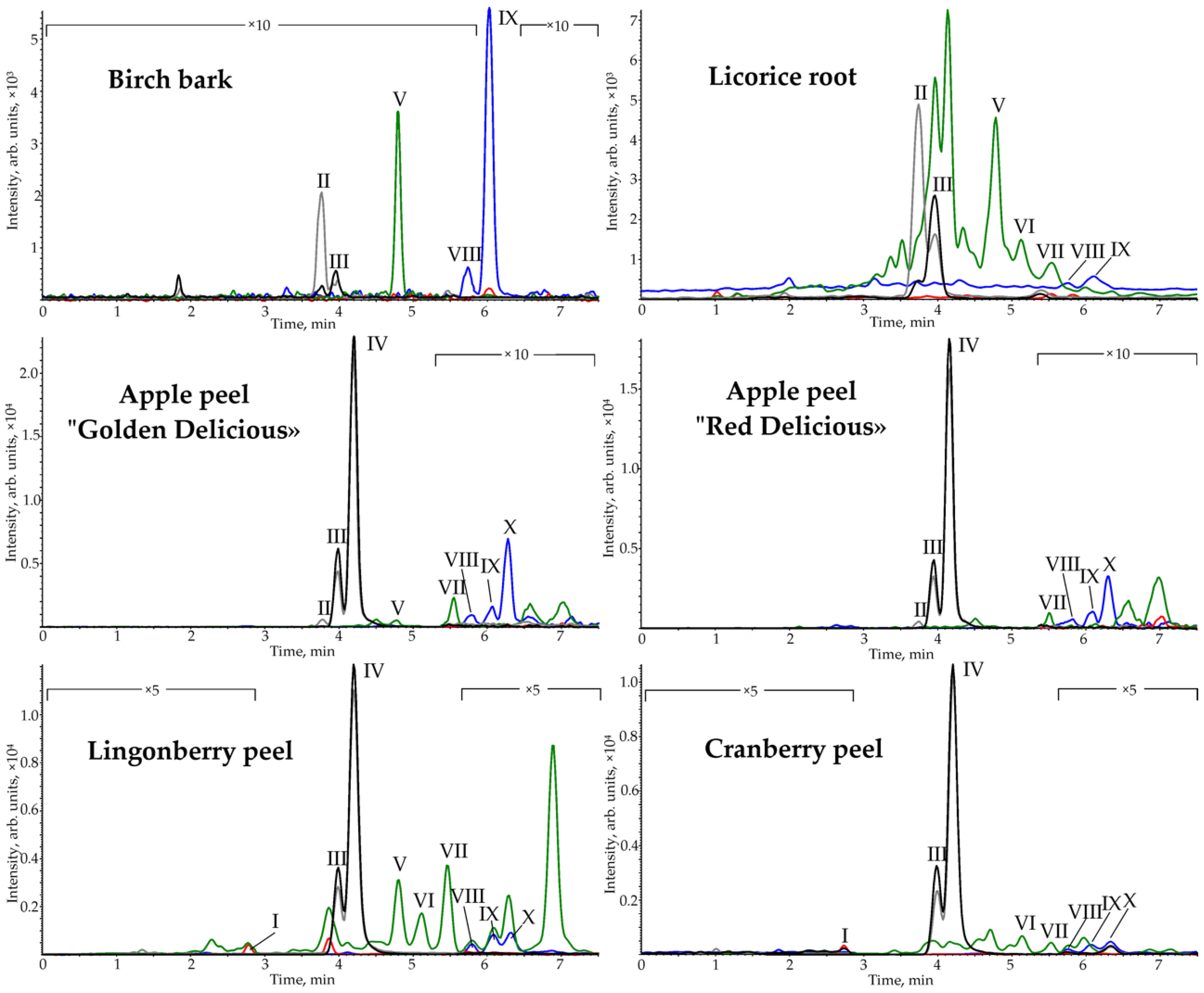
2.4. Plant Biomass Analyses
3. Materials and Methods
3.1. Reagents and Materials
3.2. Plant Materials and Extraction
3.3. Supercritical Fluid Chromatography and Mass Spectrometry
3.4. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cháirez-Ramírez, M.; Moreno-Jiménez, M.; González-Laredo, R.; Gallegos-Infante, J.; Rocha-Guzmán, N. Lupane-type triterpenes and their anti-cancer activities against most common malignant tumors: A review. EXCLI J. 2016, 15, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants–Rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–68. [Google Scholar] [CrossRef]
- Saleem, M. Lupeol, a novel anti-inflammatory and anti-cancer dietary triterpene. Cancer Lett. 2009, 285, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Alakurtti, S.; Makela, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef]
- Somova, L.O.; Nadar, A.; Rammanan, P.; Shode, F.O. Cardiovascular, antihyperlipidemic and antioxidant effects of oleanolic and ursolic acids in experimental hypertension. Phytomedicine 2003, 10, 115–121. [Google Scholar] [CrossRef]
- Mlala, S.; Oyedeji, A.O.; Gondwe, M.; Oyedeji, O.O. Ursolic Acid and Its Derivatives as Bioactive Agents. Molecules 2019, 24, 2751. [Google Scholar] [CrossRef] [Green Version]
- Jesus, J.A.; Lago, J.H.G.; Laurenti, M.D.; Yamamoto, E.S.; Passero, L.F.D. Antimicrobial activity of oleanolic and ursolic acids: An update. Evid. Based. Complement. Alternat. Med. 2015, 2015, 620472. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Sriram, D. Betulinic acid and its derivatives: A review on their biological properties. Curr. Med. Chem. 2005, 12, 657–666. [Google Scholar] [CrossRef]
- Garcia-Oliveira, P.; Otero, P.; Pereira, A.G.; Chamorro, F.; Carpena, M.; Echave, J.; Fraga-Corral, M.; Simal-Gandara, J.; Prieto, M.A. Status and challenges of plant-anticancer compounds in cancer treatment. Pharmaceuticals 2021, 14, 157. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Kannappan, R.; Aggarwal, B.B. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins 2010, 2, 2428–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, N.R.; Mandal, A.; Bhatia, D.; Siveen, K.S.; Sethi, G.; Bishayee, A. Oleanane triterpenoids in the prevention and therapy of breast cancer: Current evidence and future perspectives. Phytochem. Rev. 2014, 13, 793–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2004, 24, 90–114. [Google Scholar] [CrossRef]
- Laszczyk, M.N. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009, 75, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.N.; Ullevig, S.L.; Short, J.D.; Wang, L.; Ahn, Y.J.; Asmis, R. Ursolic acid and related analogues: Triterpenoids with broad health benefits. Antioxidants 2021, 10, 1161. [Google Scholar] [CrossRef]
- Do Nascimento, P.G.G.; Lemos, T.L.G.; Bizerra, A.M.C.; Arriaga, A.M.C.; Ferreira, D.A.; Santiago, G.M.P.; Braz-Filho, R.; Costa, J.G.M. Antibacterial and antioxidant activities of ursolic acid and derivatives. Molecules 2014, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Neto, C.C. Cranberry and blueberry: Evidence for protective effects against cancer and vascular diseases. Mol. Nutr. Food Res. 2007, 51, 652–664. [Google Scholar] [CrossRef]
- Neto, C.C. Cranberry and its phytochemicals: A review of in vitro anticancer studies. J. Nutr. 2007, 137, 186S–193S. [Google Scholar] [CrossRef] [Green Version]
- Gerhauser, C. Cancer chemopreventive potential of apples, apple juice, and apple components. Planta Med. 2008, 74, 1608–1624. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Lu, J.-J.; Huang, M.-Q.; Bao, J.-L.; Chen, X.-P.; Wang, Y.-T. Terpenoids: Natural products for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1801–1818. [Google Scholar] [CrossRef]
- He, X.; Liu, R.H. Phytochemicals of apple peels: Isolation, structure elucidation, and their antiproliferative and antioxidant activities. J. Agric. Food Chem. 2008, 56, 9905–9910. [Google Scholar] [CrossRef] [PubMed]
- Olennikov, D.N.; Vasilieva, A.G.; Chirikova, N.K. Fragaria viridis fruit metabolites: Variation of LC-MS profile and antioxidant potential during ripening and storage. Pharmaceuticals 2020, 13, 262. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz, S.; Oszmiański, J.; Wiśniewski, R. Determination of triterpenoids, carotenoids, chlorophylls, and antioxidant capacity in Allium ursinum L. at different times of harvesting and anatomical parts. Eur. Food Res. Technol. 2018, 244, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Anikeenko, E.A.; Rakhmatullina, E.N.; Falev, D.I.; Khoroshev, O.Y.; Ul’yanovskii, N.V.; Kosyakov, D.S. Application of Carbon Matrices to Screening Pentacylic Triterpenoids in Plant Feedstock by MALDI Mass Spectrometry. J. Anal. Chem. 2020, 75, 1749–1757. [Google Scholar] [CrossRef]
- Kosyakov, D.S.; Ul’yanovskii, N.V.; Falev, D.I. Determination of Triterpenoids from Birch Bark by Liquid Chromatography-Tandem Mass Spectrometry. J. Anal. Chem. 2014, 69, 50–55. [Google Scholar] [CrossRef]
- Falev, D.I.; Kosyakov, D.S.; Ul’yanovskii, N.V.; Ovchinnikov, D.V.; Shestakov, S.L. Subcritical extraction of birch bark pentacyclic triterpenes. Russ. Chem. Bull. 2017, 66, 875–881. [Google Scholar] [CrossRef]
- Falev, D.I.; Ul’yanovskii, N.V.; Ovchinnikov, D.V.; Faleva, A.V.; Kosyakov, D.S. Screening and semi-quantitative determination of pentacyclic triterpenoids in plants by liquid chromatography–tandem mass spectrometry in precursor ion scan mode. Phytochem. Anal. 2021, 32, 252–261. [Google Scholar] [CrossRef]
- Falev, D.I.; Kosyakov, D.S.; Ul’yanovskii, N.V.; Ovchinnikov, D.V. Rapid simultaneous determination of pentacyclic triterpenoids by mixed-mode liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2020, 1609, 460458. [Google Scholar] [CrossRef]
- Laboureur, L.; Ollero, M.; Touboul, D. Lipidomics by supercritical fluid chromatography. Int. J. Mol. Sci. 2015, 16, 13868–13884. [Google Scholar] [CrossRef] [Green Version]
- Tyśkiewicz, K.; Gieysztor, R.; Maziarczyk, I.; Rój, E.; Skalicka-Woźniak, K. Supercritical fluid chromatography with photodiode array detection in the determination of fat-soluble vitamins in hemp seed oil and waste fish oil. Molecules 2018, 23, 1131. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikov, D.V.; Pokrovskiy, O.I.; Kosyakov, D.S.; Bogolitsyn, K.G.; Ul’yanovskii, N.V.; Falev, D.I. Evaluation of temperature and pressure effects on retention in supercritical fluid chromatography on polar stationary phases. J. Chromatogr. A 2020, 1610, 460600. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; D’Orazio, G.; Gentili, A.; Fanali, S. Analysis of enantiomers in products of food interest. Molecules 2019, 24, 1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesellier, E.; Destandau, E.; Grigoras, C.; Fougère, L.; Elfakir, C. Fast separation of triterpenoids by supercritical fluid chromatography/evaporative light scattering detector. J. Chromatogr. A 2012, 1268, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Fukui, H.; Tabata, M. Examination of triterpenoids produced by callus and cell suspension cultures of Glycyrrhiza glabra. Plant Cell Rep. 1988, 7, 508–511. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Zhang, H.; Yue, J.; Sun, Y.; Zhang, X.; Zhao, Y. Synthesis of triterpenoid derivatives and their anti-tumor and anti-hepatic fibrosis activities. Nat. Prod. Res. 2020, 34, 766–772. [Google Scholar] [CrossRef]
- Jemmali, Z.; Chartier, A.; Dufresne, C.; Elfakir, C. Optimization of the derivatization protocol of pentacyclic triterpenes prior to their gas chromatography-mass spectrometry analysis in plant extracts. Talanta 2016, 147, 35–43. [Google Scholar] [CrossRef]
- De la Peña Armada, R.; Bronze, M.R.; Matias, A.; Mateos-Aparicio, I. Triterpene-Rich Supercritical CO2 Extracts from Apple By-product Protect Human Keratinocytes Against ROS. Food Bioprocess Technol. 2021, 14, 909–919. [Google Scholar] [CrossRef]
- Poirier, B.C.; Buchanan, D.A.; Rudell, D.R.; Mattheis, J.P. Differential Partitioning of Triterpenes and Triterpene Esters in Apple Peel. J. Agric. Food Chem. 2018, 66, 1800–1806. [Google Scholar] [CrossRef]
- Szakiel, A.; Pa̧czkowski, C.; Koivuniemi, H.; Huttunen, S. Comparison of the triterpenoid content of berries and leaves of lingonberry Vaccinium vitis-idaea from Finland and Poland. J. Agric. Food Chem. 2012, 60, 4994–5002. [Google Scholar] [CrossRef]
- Szakiel, A.; Pączkowski, C.; Pensec, F.; Bertsch, C. Fruit cuticular waxes as a source of biologically active triterpenoids. Phytochem. Rev. 2012, 11, 263–284. [Google Scholar] [CrossRef] [Green Version]
- Klavins, L.; Klavins, M. Cuticular wax composition of wild and cultivated northern berries. Foods 2020, 9, 587. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Monoisotopic Mass, Da | Precursor Ion, m/z | Product Ion, m/z | Declustering Potential, V | Entrance Potential, V | Collision Energy, eV |
|---|---|---|---|---|---|---|
| I | 426 | 427 | 95 | 41 | 4.5 | 45 |
| II | 456 | 439 | 95 | 61 | 5.5 | 45 |
| III | 456 | 439 | 191 | 43 | 5.0 | 19 |
| IV | 456 | 439 | 191 | 43 | 6.0 | 49 |
| V | 426 | 409 | 95 | 55 | 6.5 | 47 |
| VI | 426 | 409 | 95 | 55 | 8.0 | 49 |
| VII | 426 | 409 | 95 | 55 | 7.5 | 59 |
| VIII | 442 | 425 | 95 | 50 | 5.5 | 47 |
| IX | 442 | 425 | 95 | 50 | 6.0 | 19 |
| X | 442 | 425 | 95 | 50 | 7.5 | 21 |
| Analyte | APPI | APCI | ||||||
|---|---|---|---|---|---|---|---|---|
| Linear Concentration Range, μg·L−1 | a | R2 | LOQ, μg·L−1 | Linear Concentration Range, μg·L−1 | a | R2 | LOQ, μg·L−1 | |
| I | 33–2000 | 6.163 | 0.99288 | 33 | 20–2000 | 26.076 | 0.99945 | 20 |
| II | 7.0–2000 | 50.235 | 0.99278 | 7.0 | 11–2000 | 87.018 | 0.99997 | 11 |
| III | 3.5–1000 | 42.627 | 0.99422 | 3.5 | 4.0–1000 | 75.104 | 0.99997 | 4.0 |
| IV | 4.6–1000 | 38.455 | 0.99033 | 4.6 | 4.7–1000 | 68.122 | 0.99994 | 4.7 |
| V | 3.8–1000 | 139.33 | 0.99283 | 3.8 | 2.6–1000 | 219.15 | 0.99998 | 2.6 |
| VI | 4.5–1000 | 119.78 | 0.99833 | 4.5 | 2.7–1000 | 211.91 | 0.99997 | 2.7 |
| VII | 3.8–1000 | 147.89 | 0.99243 | 3.8 | 2.3–1000 | 253.00 | 0.99996 | 2.3 |
| VIII | 20–2000 | 35.314 | 0.99206 | 20 | 9.8–2000 | 92.152 | 0.99995 | 9.8 |
| IX | 11–1000 | 68.244 | 0.99116 | 11 | 5.5–1000 | 175.25 | 0.99997 | 5.5 |
| X | 27–2000 | 27.677 | 0.99410 | 27 | 13–2000 | 75.618 | 0.99993 | 13 |
| Analyte | Birch Bark | Licorice Root | Apple Peel “Golden Delicious” | Apple Peel “Red Delicious” | Lingonberry Peel | Cranberry Peel |
|---|---|---|---|---|---|---|
| I | - | - | - | - | 0.17 ± 0.01 | 0.14 ± 0.01 |
| II | 13 ± 1 | 0.16 ± 0.01 | 0.79 ± 0.01 | 0.47 ± 0.02 | - | - |
| III | 3.6 ± 0.5 | 0.10 ± 0.01 | 10 ± 1 | 6.3 ± 0.5 | 3.5 ± 0.3 | 3.2 ± 0.2 |
| IV | - | - | 49 ± 5 | 32 ± 1 | 15 ± 2 | 14 ± 1 |
| V | 4.6 ± 0.3 | 0.043 ± 0.001 | 0.26 ± 0.02 | - | 0.80 ± 0.01 | - |
| VI | - | 0.0076 ± 0.0008 | - | - | 0.58 ± 0.01 | 0.23 ± 0.01 |
| VII | - | 0.0062 ± 0.0001 | 0.088 ± 0.006 | 0.027 ± 0.001 | 0.84 ± 0.01 | 0.12 ± 0.01 |
| VIII | 2.9 ± 0.1 | 0.0041 ± 0.0001 | 0.16 ± 0.01 | 0.090 ± 0.008 | 0.058 ± 0.003 | 0.030 ± 0.003 |
| IX | 250 ± 10 | 0.0073 ± 0.0002 | 0.12 ± 0.02 | 0.077 ± 0.002 | 0.072 ± 0.004 | 0.022 ± 0.002 |
| X | - | - | 1.1 ± 0.1 | 0.48 ± 0.05 | 0.17 ± 0.01 | 0.096 ± 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falev, D.I.; Ovchinnikov, D.V.; Voronov, I.S.; Faleva, A.V.; Ul’yanovskii, N.V.; Kosyakov, D.S. Supercritical Fluid Chromatography—Tandem Mass Spectrometry for Rapid Quantification of Pentacyclic Triterpenoids in Plant Extracts. Pharmaceuticals 2022, 15, 629. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050629
Falev DI, Ovchinnikov DV, Voronov IS, Faleva AV, Ul’yanovskii NV, Kosyakov DS. Supercritical Fluid Chromatography—Tandem Mass Spectrometry for Rapid Quantification of Pentacyclic Triterpenoids in Plant Extracts. Pharmaceuticals. 2022; 15(5):629. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050629
Chicago/Turabian StyleFalev, Danil I., Denis V. Ovchinnikov, Ilya S. Voronov, Anna V. Faleva, Nikolay V. Ul’yanovskii, and Dmitry S. Kosyakov. 2022. "Supercritical Fluid Chromatography—Tandem Mass Spectrometry for Rapid Quantification of Pentacyclic Triterpenoids in Plant Extracts" Pharmaceuticals 15, no. 5: 629. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050629