
Structure-Based Optimization of 1,2,4-Triazole-3-Thione Derivatives: Improving Inhibition of NDM-/VIM-Type Metallo-β-Lactamases and Synergistic Activity on Resistant Bacteria
 , , , , , , , , ,
, , , , , , , , ,  , and
, and
Abstract
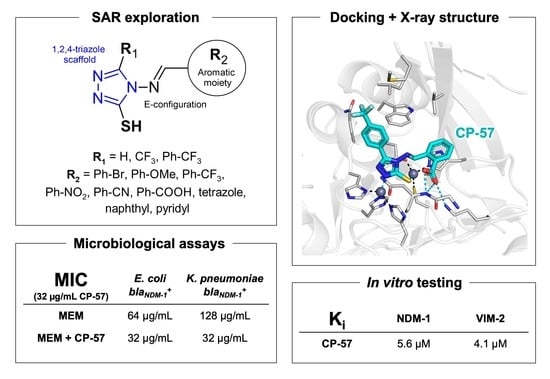
:
1. Introduction
2. Results and Discussion
2.1. Molecular Interaction Field (MIF) Analysis
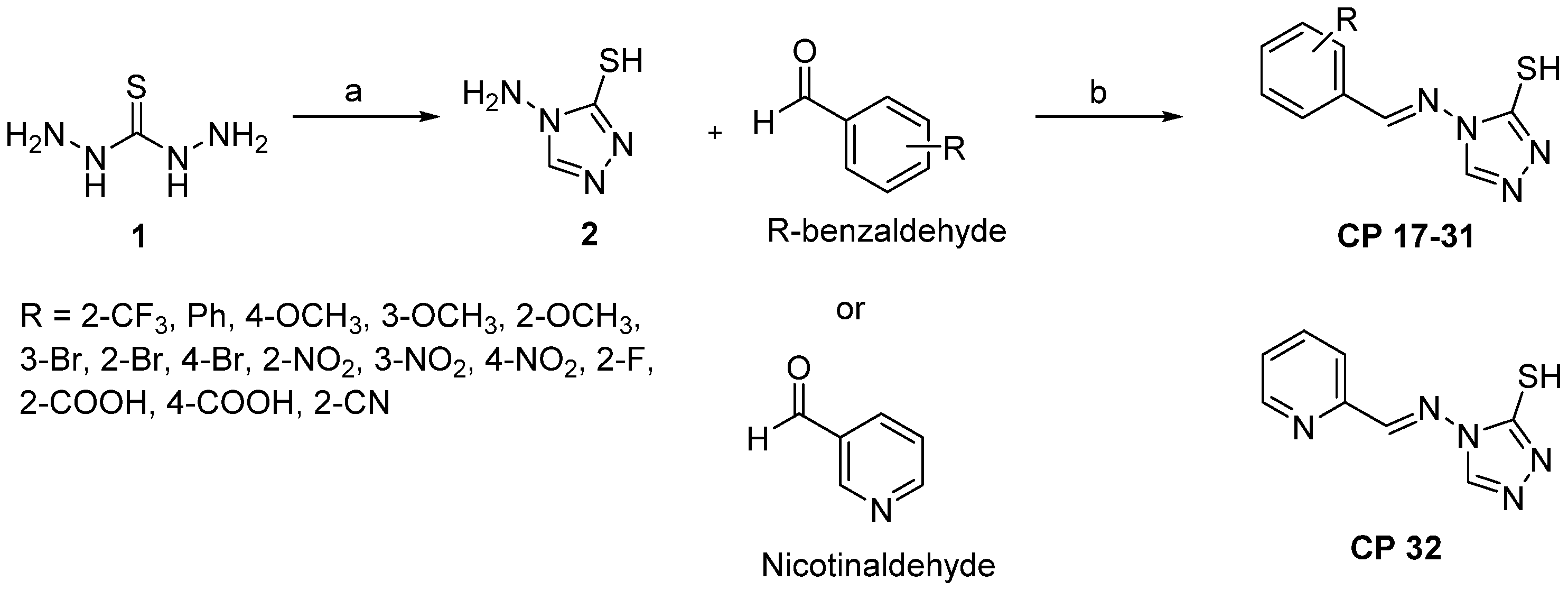
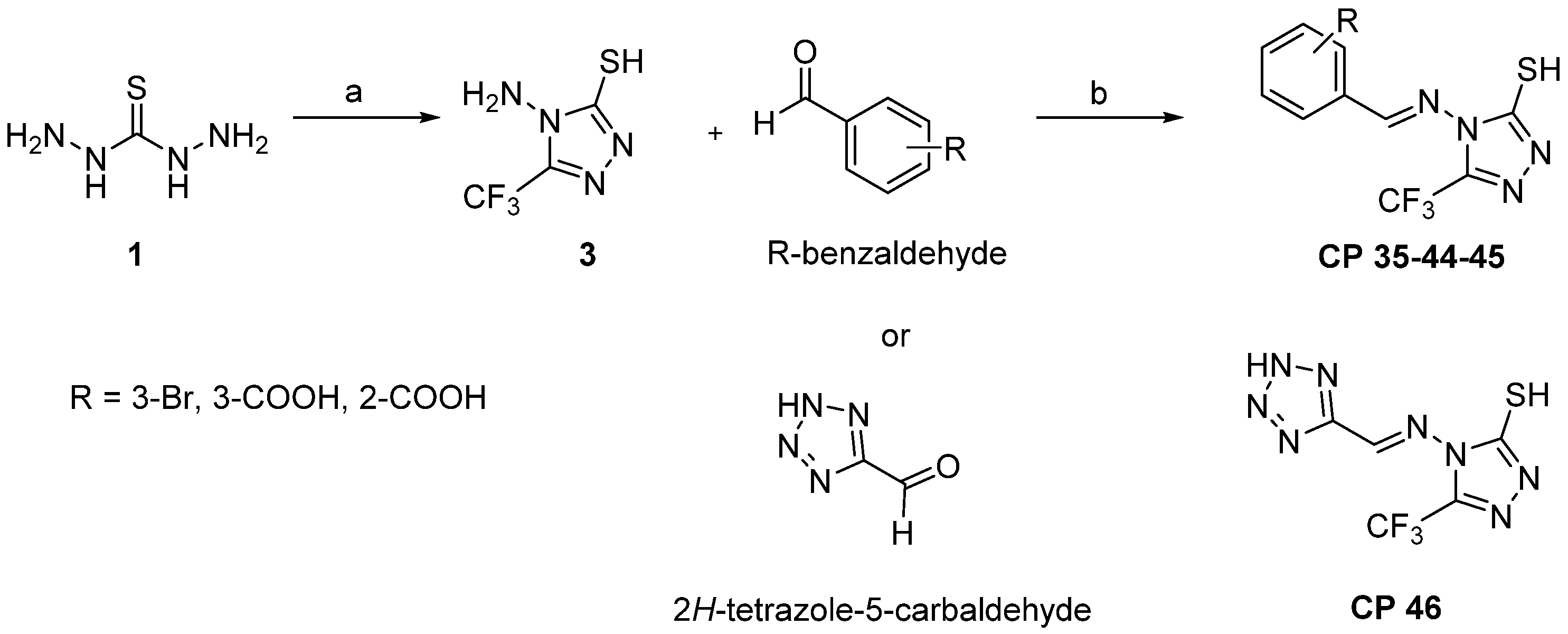
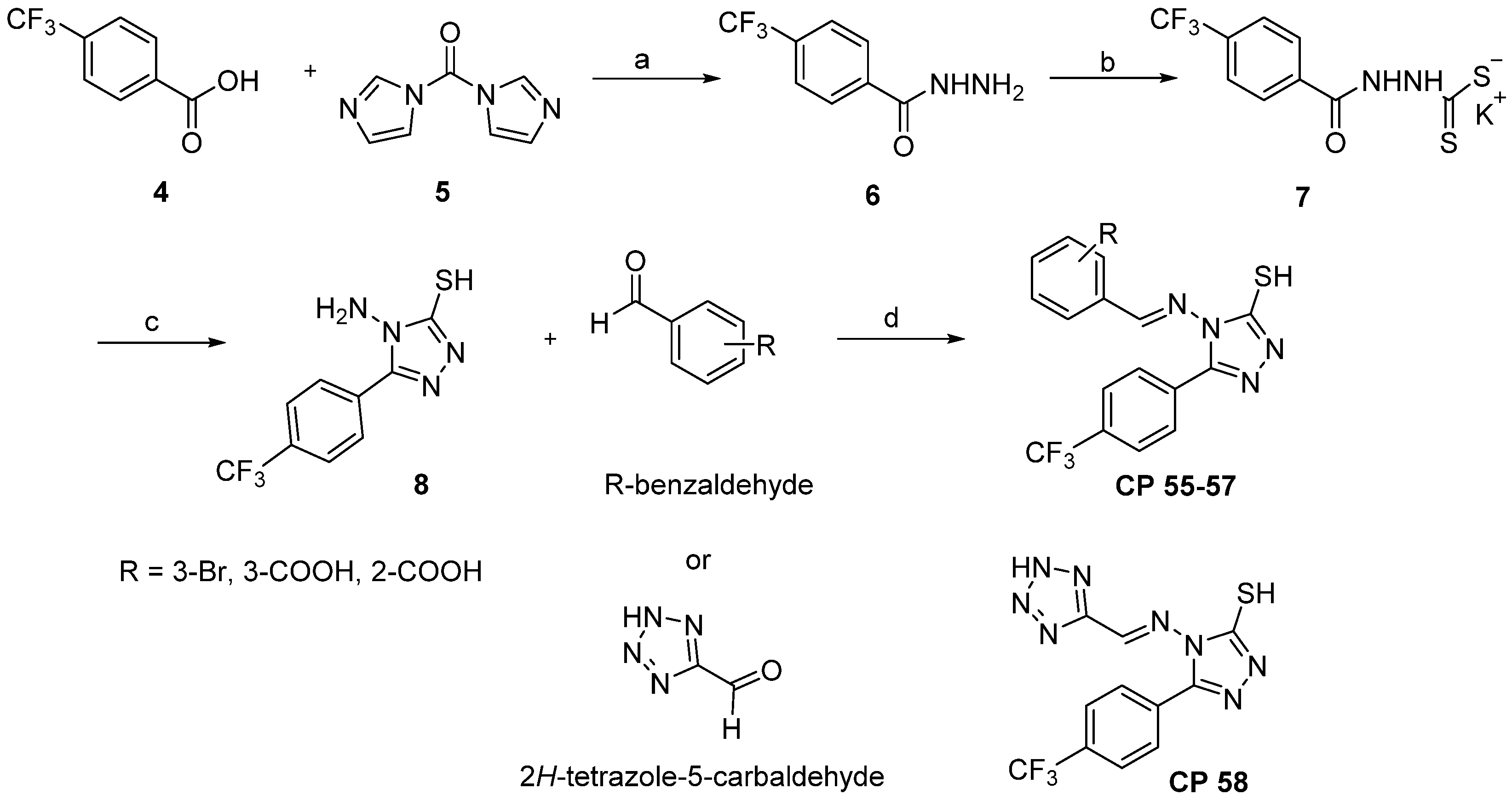
2.2. Chemistry
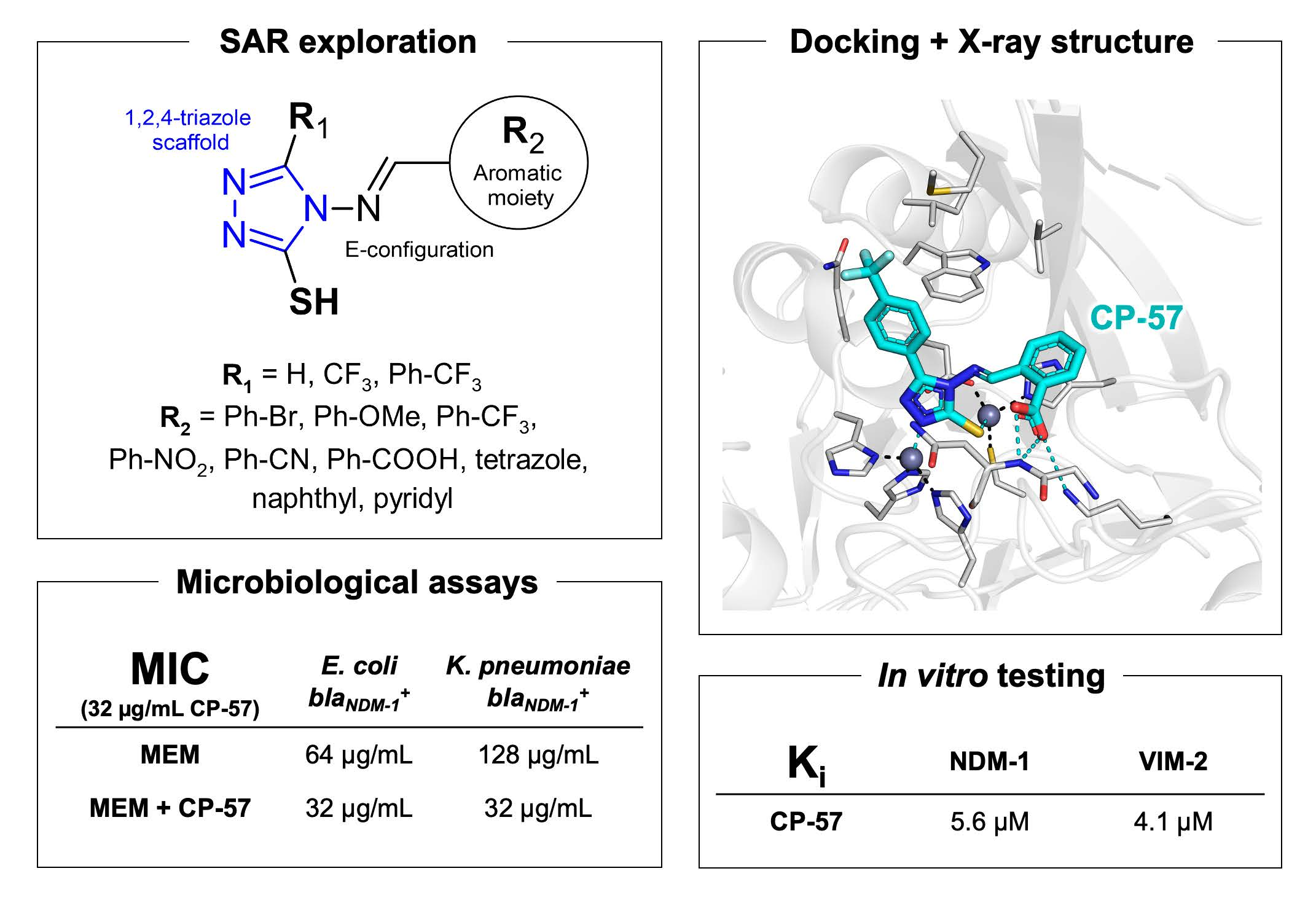
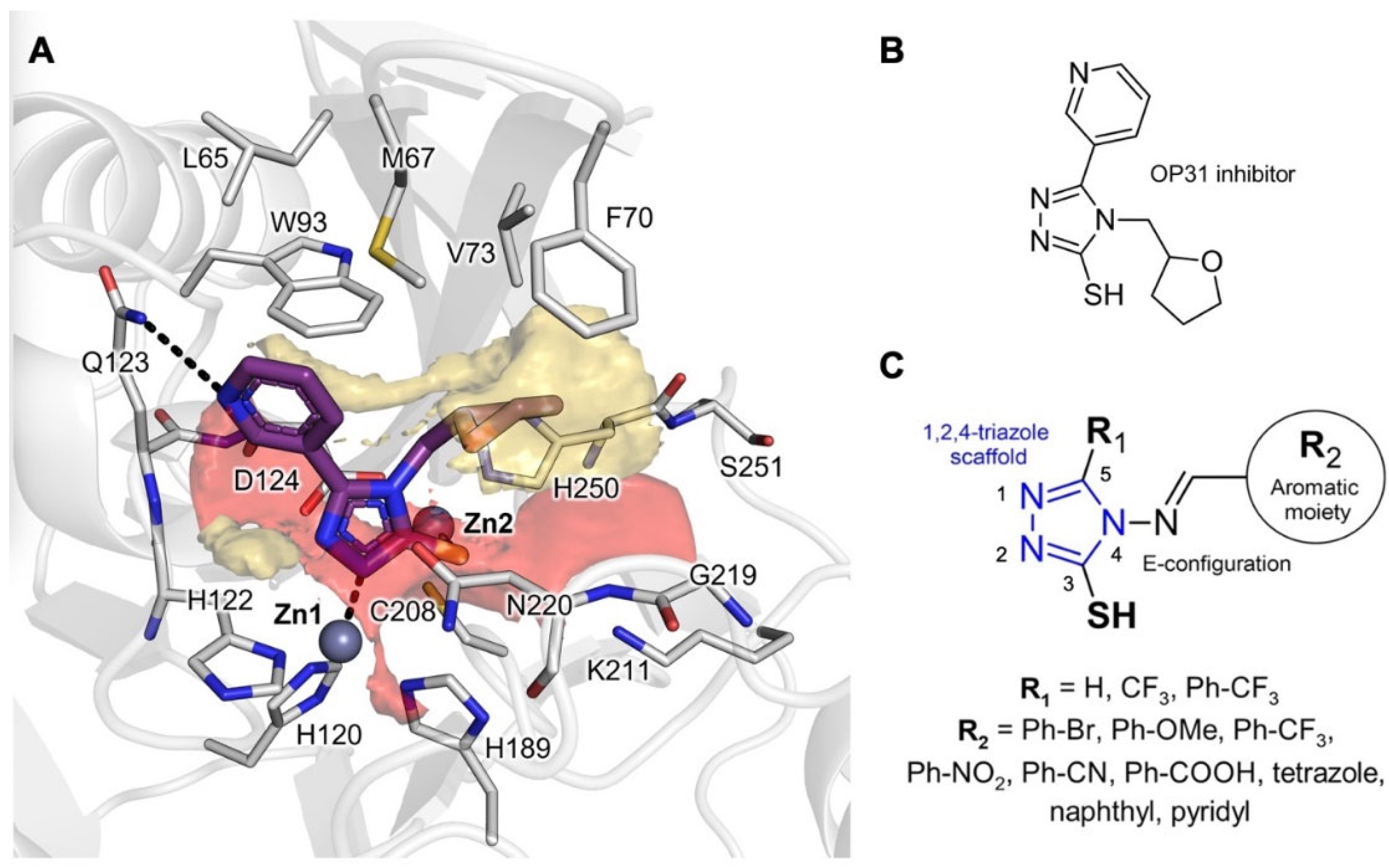
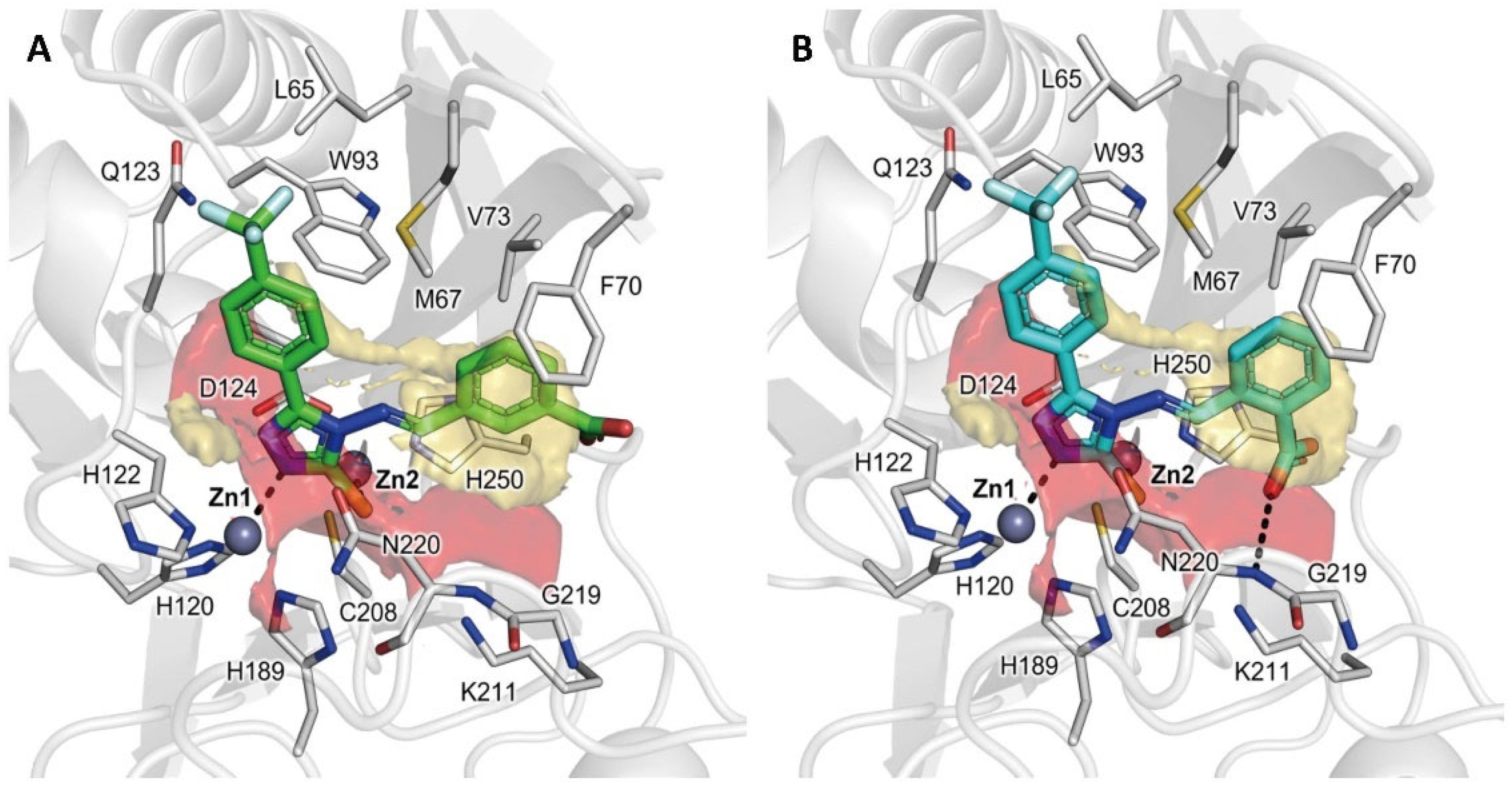
2.3. Molecular Modelling
2.4. In Vitro Enzyme Inhibition Assays
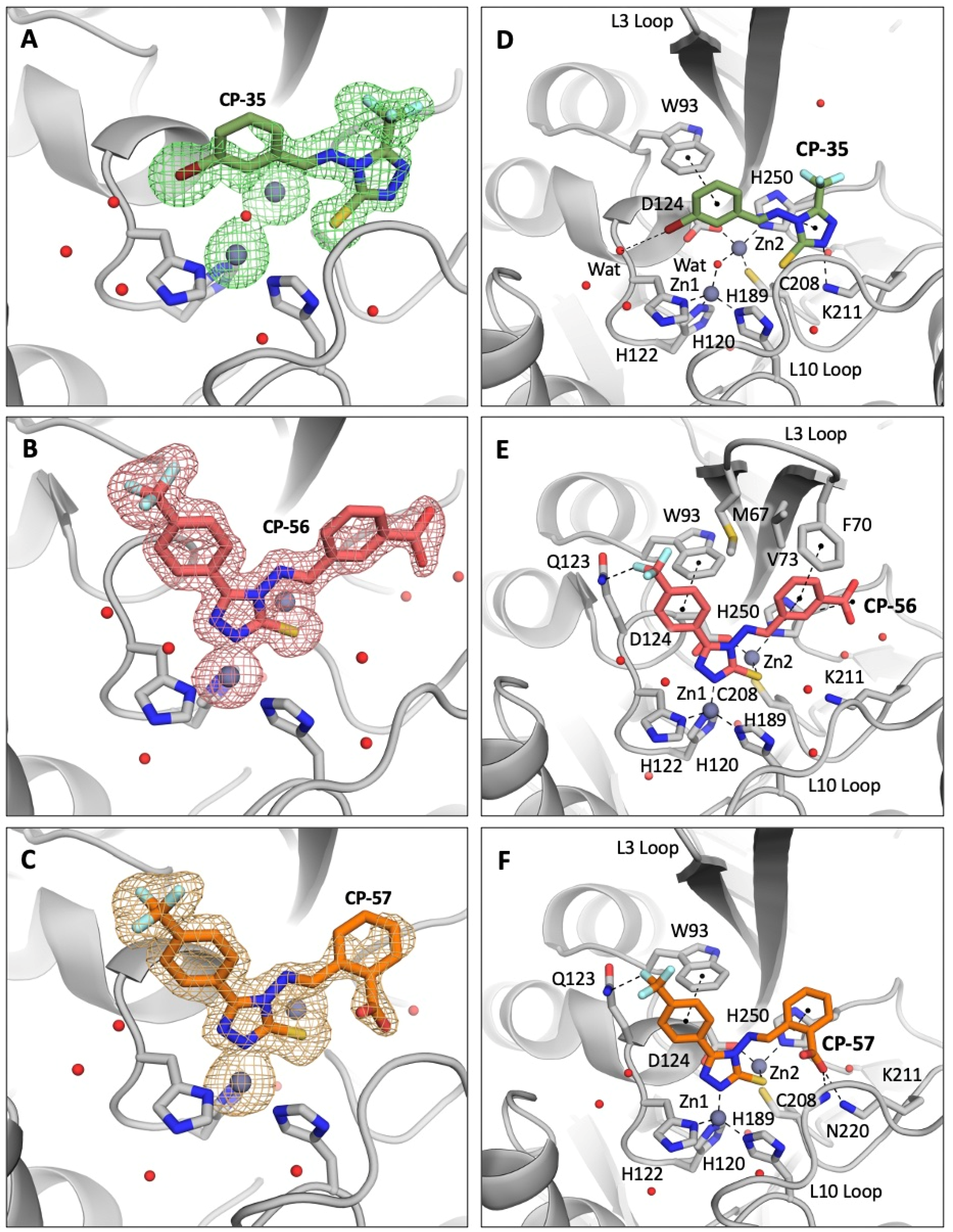
2.5. X-ray Crystallography
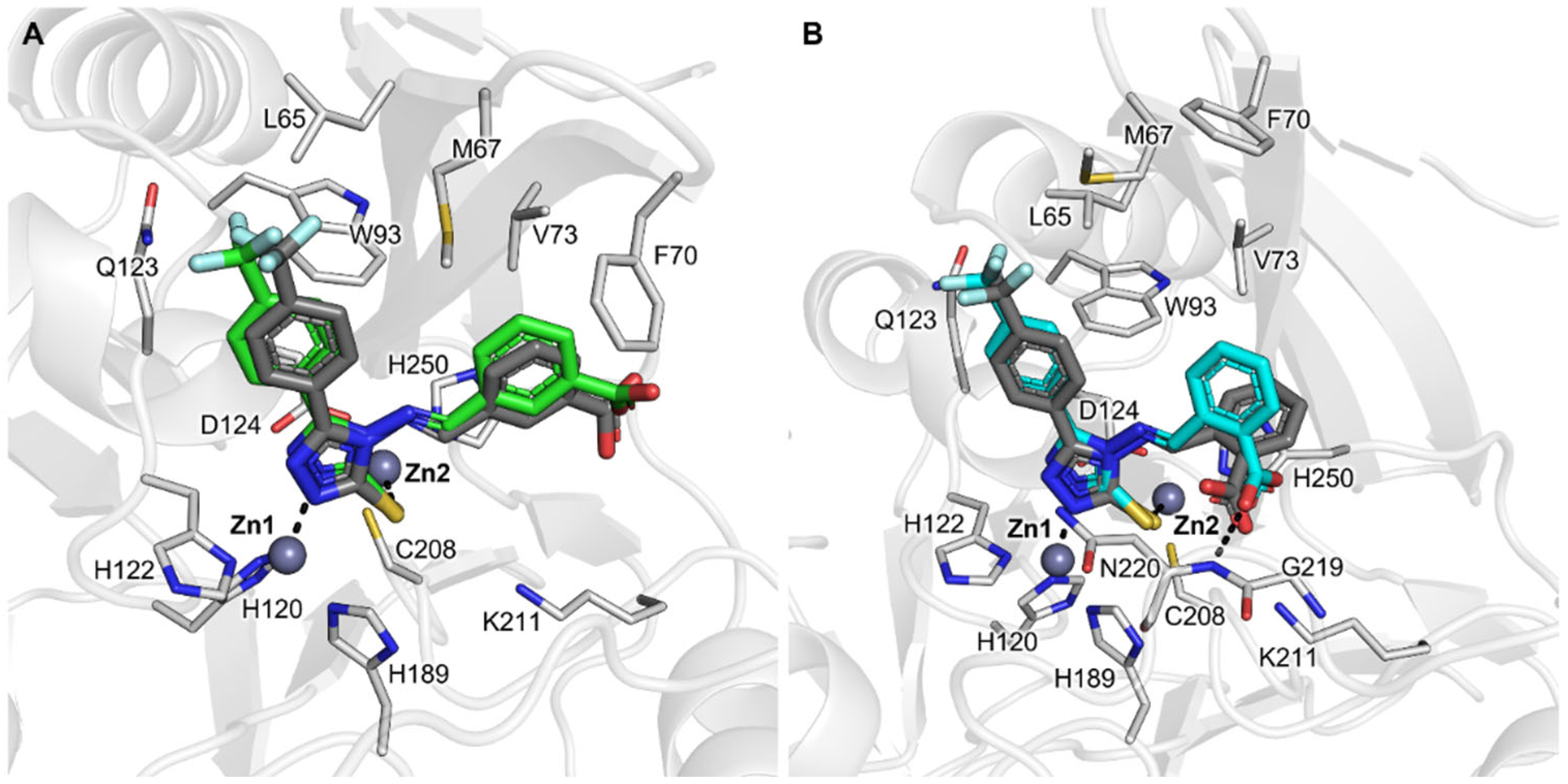
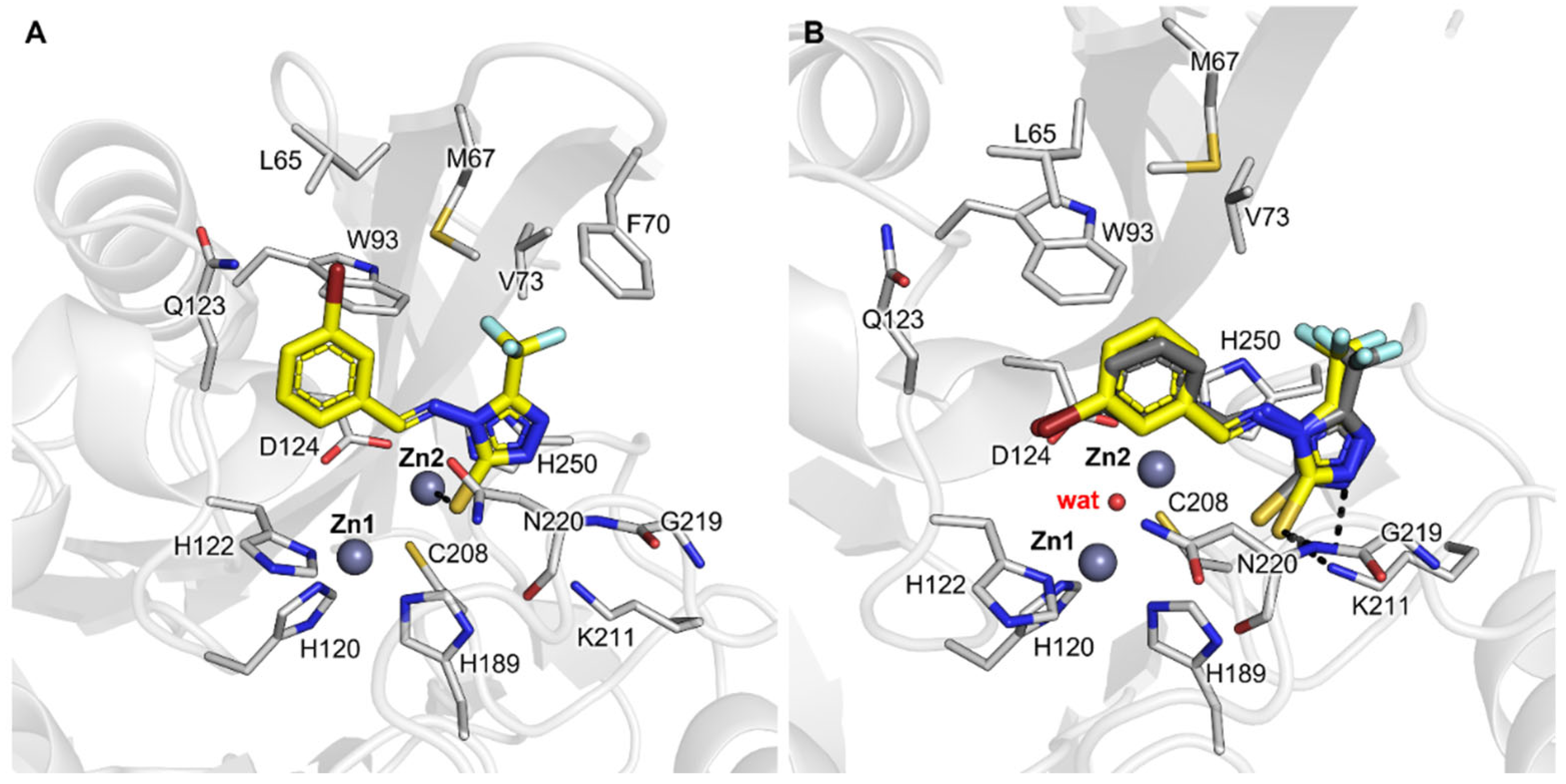
2.6. Assessment and Refinement of Docking Results
2.7. In Vitro Antibacterial Synergistic Activity
3. Materials and Methods
3.1. MIF Analysis for Inhibitor Design and Molecular Docking
3.2. Chemistry
3.3. In Vitro Enzyme Inhibition and Microbiological Assays
3.4. NDM-1 Overexpression, Purification, and Crystallization in Complex with Inhibitors
3.5. Crystallography-Structure Building and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Venter, H. Reversing Resistance to Counter Antimicrobial Resistance in the World Health Organisation’s Critical Priority of Most Dangerous Pathogens. Biosci. Rep. 2019, 39, BSR20180474. [Google Scholar] [CrossRef]
- Global Action Plan on Antimicrobial Resistance. Available online: https://www.who.int/publications-detail-redirect/9789241509763 (accessed on 12 August 2023).
- Walsh, T.R.; Toleman, M.A. The Emergence of Pan-Resistant Gram-Negative Pathogens Merits a Rapid Global Political Response. J. Antimicrob. Chemother. 2012, 67, 1–3. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Exner, M.; Bhattacharya, S.; Christiansen, B.; Gebel, J.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Kramer, A.; Larson, E.; et al. Antibiotic Resistance: What Is so Special about Multidrug-Resistant Gram-Negative Bacteria? GMS Hyg. Infect. Control 2017, 12, Doc05. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016. [Google Scholar]
- Cho, H.; Uehara, T.; Bernhardt, T.G. Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yan, Y.-H.; Schofield, C.J.; McNally, A.; Zong, Z.; Li, G.-B. Metallo-β-Lactamase-Mediated Antimicrobial Resistance and Progress in Inhibitor Discovery. Trends Microbiol. 2023, 31, 735–748. [Google Scholar] [CrossRef]
- Bahr, G.; González, L.J.; Vila, A.J. Metallo-β-Lactamases in the Age of Multidrug Resistance: From Structure and Mechanism to Evolution, Dissemination, and Inhibitor Design. Chem. Rev. 2021, 121, 7957–8094. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Naas, T.; Poirel, L. Global Spread of Carbapenemase-Producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef]
- Rolain, J.M.; Parola, P.; Cornaglia, G. New Delhi Metallo-Beta-Lactamase (NDM-1): Towards a New Pandemia? Clin. Microbiol. Infect. 2010, 16, 1699–1701. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-Negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef]
- Martin, M.J.; Corey, B.W.; Sannio, F.; Hall, L.R.; MacDonald, U.; Jones, B.T.; Mills, E.G.; Harless, C.; Stam, J.; Maybank, R.; et al. Anatomy of an Extensively Drug-Resistant Klebsiella Pneumoniae Outbreak in Tuscany, Italy. Proc. Natl. Acad. Sci. USA 2021, 118, e2110227118. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-Membrane Vesicles from Gram-Negative Bacteria: Biogenesis and Functions. Nat. Rev. Microbiol. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- González, L.J.; Bahr, G.; González, M.M.; Bonomo, R.A.; Vila, A.J. In-Cell Kinetic Stability Is an Essential Trait in Metallo-β-Lactamase Evolution. Nat. Chem. Biol. 2023, 19, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- González, L.J.; Bahr, G.; Nakashige, T.G.; Nolan, E.M.; Bonomo, R.A.; Vila, A.J. Membrane Anchoring Stabilizes and Favors Secretion of New Delhi Metallo-β-Lactamase. Nat. Chem. Biol. 2016, 12, 516–522. [Google Scholar] [CrossRef]
- Poirel, L.; Bonnin, R.A.; Nordmann, P. Analysis of the Resistome of a Multidrug-Resistant NDM-1-Producing Escherichia Coli Strain by High-Throughput Genome Sequencing. Antimicrob. Agents Chemother. 2011, 55, 4224–4229. [Google Scholar] [CrossRef]
- Rogers, B.A.; Sidjabat, H.E.; Silvey, A.; Anderson, T.L.; Perera, S.; Li, J.; Paterson, D.L. Treatment Options for New Delhi Metallo-Beta-Lactamase-Harboring Enterobacteriaceae. Microb. Drug Resist. 2013, 19, 100–103. [Google Scholar] [CrossRef]
- Drekonja, D.M.; Beekmann, S.E.; Elliott, S.; Mukundan, D.; Polenakovik, H.; Rosenthal, M.E.; Tamma, P.D.; Polgreen, P.M.; Weissman, S.J. Challenges in the Management of Infections Due to Carbapenem-Resistant Enterobacteriaceae. Infect. Control Hosp. Epidemiol. 2014, 35, 437–439. [Google Scholar] [CrossRef]
- Giamarellou, H.; Karaiskos, I. Current and Potential Therapeutic Options for Infections Caused by Difficult-to-Treat and Pandrug Resistant Gram-Negative Bacteria in Critically Ill Patients. Antibiotics 2022, 11, 1009. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten Years with New Delhi Metallo-β-Lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Vella, P.; Hussein, W.M.; Leung, E.W.W.; Clayton, D.; Ollis, D.L.; Mitić, N.; Schenk, G.; McGeary, R.P. The Identification of New Metallo-β-Lactamase Inhibitor Leads from Fragment-Based Screening. Bioorg. Med. Chem. Lett. 2011, 21, 3282–3285. [Google Scholar] [CrossRef]
- Christopeit, T.; Carlsen, T.J.O.; Helland, R.; Leiros, H.-K.S. Discovery of Novel Inhibitor Scaffolds against the Metallo-β-Lactamase VIM-2 by Surface Plasmon Resonance (SPR) Based Fragment Screening. J. Med. Chem. 2015, 58, 8671–8682. [Google Scholar] [CrossRef]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.-D.; Cendron, L.; et al. Virtual Screening Identifies Broad-Spectrum β-Lactamase Inhibitors with Activity on Clinically Relevant Serine- and Metallo-Carbapenemases. Sci. Rep. 2020, 10, 12763. [Google Scholar] [CrossRef]
- Verdirosa, F.; Gavara, L.; Sevaille, L.; Tassone, G.; Corsica, G.; Legru, A.; Feller, G.; Chelini, G.; Mercuri, P.S.; Tanfoni, S.; et al. 1,2,4-Triazole-3-Thione Analogues with a 2-Ethylbenzoic Acid at Position 4 as VIM-Type Metallo-β-Lactamase Inhibitors. ChemMedChem 2022, 17, e202100699. [Google Scholar] [CrossRef] [PubMed]
- Legru, A.; Verdirosa, F.; Vo-Hoang, Y.; Tassone, G.; Vascon, F.; Thomas, C.A.; Sannio, F.; Corsica, G.; Benvenuti, M.; Feller, G.; et al. Optimization of 1,2,4-Triazole-3-Thiones toward Broad-Spectrum Metallo-β-Lactamase Inhibitors Showing Potent Synergistic Activity on VIM- and NDM-1-Producing Clinical Isolates. J. Med. Chem. 2022, 65, 16392–16419. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Gianquinto, E.; Montanari, M.; Maso, L.; Bellio, P.; Cebrián-Sastre, E.; Celenza, G.; Blázquez, J.; Cendron, L.; Spyrakis, F.; et al. 4-Amino-1,2,4-Triazole-3-Thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases. Pharmaceuticals 2020, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, A.R.; Beltz, J.; King, E.; Ercal, N. Medicinal Thiols: Current Status and New Perspectives. Mini-Rev. Med. Chem. 2020, 20, 513–529. [Google Scholar] [CrossRef]
- Murphy, B.; Das, B.; Wei, C.; Li, L. Hck Inhibitors for the Treatment of Fibrosis and Cancer, WO2020/205921. 2020. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020205921 (accessed on 12 August 2023).
- Chen, M.; Wang, X.-F.; Wang, S.-S.; Feng, Y.-X.; Chen, F.; Yang, C.-L. Synthesis, Characterization and Fungicidal Activities of Novel Fluorinated 3,5-Disubstituted-4H-1,2,4-Triazol-4-Amines. J. Fluor. Chem. 2012, 135, 323–329. [Google Scholar] [CrossRef]
- Gavara, L.; Sevaille, L.; De Luca, F.; Mercuri, P.; Bebrone, C.; Feller, G.; Legru, A.; Cerboni, G.; Tanfoni, S.; Baud, D.; et al. 4-Amino-1,2,4-Triazole-3-Thione-Derived Schiff Bases as Metallo-β-Lactamase Inhibitors. Eur. J. Med. Chem. 2020, 208, 112720. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- King, D.; Strynadka, N. Crystal Structure of New Delhi Metallo-β-Lactamase Reveals Molecular Basis for Antibiotic Resistance. Protein Sci. 2011, 20, 1484–1491. [Google Scholar] [CrossRef]
- Sun, Z.; Hu, L.; Sankaran, B.; Prasad, B.V.V.; Palzkill, T. Differential Active Site Requirements for NDM-1 β-Lactamase Hydrolysis of Carbapenem versus Penicillin and Cephalosporin Antibiotics. Nat. Commun. 2018, 9, 4524. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.-D.; Lamotte-Brasseur, J.; Galleni, M.; Amicosante, G.; Frère, J.-M.; Rossolini, G.M. On Functional and Structural Heterogeneity of VIM-Type Metallo-β-Lactamases. J. Antimicrob. Chemother. 2003, 51, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Mojica, M.F.; Mahler, S.G.; Bethel, C.R.; Taracila, M.A.; Kosmopoulou, M.; Papp-Wallace, K.M.; Llarrull, L.I.; Wilson, B.M.; Marshall, S.H.; Wallace, C.J.; et al. Exploring the Role of Residue 228 in Substrate and Inhibitor Recognition by VIM Metallo-β-Lactamases. Biochemistry 2015, 54, 3183–3196. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Rc, W.; Pj, G. The Role of Hydrogen-Bonds in Drug Binding. Progress. Clin. Biol. Res. 1989, 289, 433–444. [Google Scholar]
- Sciabola, S.; Baroni, M.; Carosati, E.; Cruciani, G. Recent Improvements in the GRID Force Field. 1. The Docking Procedure GLUE. In QSAR and Molecular Modelling in Rational Design of Bioactive Molecules; CADDD Society: Ankara, Turkey, 2005; pp. 47–49. ISBN 9750078209. [Google Scholar]
- Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Tamilselvi, A.; Mugesh, G. Metallo-β-Lactamase-Catalyzed Hydrolysis of Cephalosporins: Some Mechanistic Insights into the Effect of Heterocyclic Thiones on Enzyme Activity. Inorg. Chem. 2011, 50, 749–756. [Google Scholar] [CrossRef]
- Nauton, L.; Kahn, R.; Garau, G.; Hernandez, J.F.; Dideberg, O. Structural Insights into the Design of Inhibitors for the L1 Metallo-Beta-Lactamase from Stenotrophomonas Maltophilia. J. Mol. Biol. 2008, 375, 257–269. [Google Scholar] [CrossRef]
- Klingler, F.-M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Müller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M.; et al. Approved Drugs Containing Thiols as Inhibitors of Metallo-β-Lactamases: Strategy to Combat Multidrug-Resistant Bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef]
- Büttner, D.; Kramer, J.S.; Klingler, F.-M.; Wittmann, S.K.; Hartmann, M.R.; Kurz, C.G.; Kohnhäuser, D.; Weizel, L.; Brüggerhoff, A.; Frank, D.; et al. Challenges in the Development of a Thiol-Based Broad-Spectrum Inhibitor for Metallo-β-Lactamases. ACS Infect. Dis. 2018, 4, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Green, N.; Hu, Y.; Janz, K.; Li, H.-Q.; Kaila, N.; Guler, S.; Thomason, J.; Joseph-McCarthy, D.; Tam, S.Y.; Hotchandani, R.; et al. Inhibitors of Tumor Progression Loci-2 (Tpl2) Kinase and Tumor Necrosis Factor α (TNF-α) Production: Selectivity and in Vivo Antiinflammatory Activity of Novel 8-Substituted-4-anilino-6-aminoquinoline-3-carbonitriles. Journal of Medicinal Chemistry 2007, 50(19), 4728–4745. [Google Scholar] [CrossRef] [PubMed]
- M07: Dilution AST for Aerobically Grown Bacteria—CLSI. Available online: https://clsi.org/standards/products/microbiology/documents/m07/ (accessed on 12 August 2023).
- Feng, B.Y.; Shoichet, B.K. A Detergent-Based Assay for the Detection of Promiscuous Inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Long, F.; Nicholls, R.A.; Emsley, P.; Gražulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A Stereochemical Description Generator for Ligands. Acta Crystallogr. D Struct. Biol. 2017, 73, 112–122. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein–Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Research 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.C.; Docquier, J.-D.; Uehara, T.; Myers, C.L.; Six, D.A.; Chatwin, C.L.; John, K.J.; Vernacchio, S.F.; Cusick, S.M.; Trout, R.E.L.; et al. VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-β-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2020, 64, e01963-19. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Rubio-Aparicio, D.; Sun, D.; Dudley, M.; Lomovskaya, O. In Vitro Activity of the Ultrabroad-Spectrum-Beta-Lactamase Inhibitor QPX7728 against Carbapenem-Resistant Enterobacterales with Varying Intrinsic and Acquired Resistance Mechanisms. Antimicrob. Agents Chemother. 2020, 64, e00757-20. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | NDM-1 IC50 (μM) |
|---|---|---|
| CP 17 |  | 226 ± 26 |
| CP 18 |  | 52 ± 7.5 |
| CP 22 |  | 88.5 ± 3.7 (Ki of 42.7 a ± 2.1) |
| CP 23 |  | 83.0 ± 3.3 |
| CP 24 |  | 83.1 ± 2.7 |
| CP 29 |  | NI b |
| CP 30 |  | NI b |
| Compound | Structure | NDM-1 Ki (μM) | VIM-2 Ki (μM) |
|---|---|---|---|
| CP 35 |  | 25.8 a ± 0.7 | 22.6 b ± 1.1 |
| CP 44 |  | 115 b ± 20 | 22.3 b ±1.1 |
| CP 45 |  | 83 b ± 4.1 | NI |
| CP 46 |  | 70 b ± 3.2 | 215 b ± 10 |
| CP 55 |  | 220 b ± 11 | 40 b ± 2 |
| CP 56 |  | 24.5 ± 0.5 | 13.8 b ± 1.6 |
| CP 57 |  | 5.6 ± 0.8 | 4.1 ± 0.8 |
| CP 58 |  | 78 b ± 8 | 28 b ± 3.2 |
| Cpd (32 μg/mL) | MEM MIC (μg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| E. coli MO-287 (NDM-1) | E. coli MO-978 (NDM1) | K. pneumoniae SI-1072 (NDM-1) | K. pneumoniae SI-518 (NDM-1) | K. pneumoniae VA-416/02 (VIM-4) | P. aeruginosa VA-182/00 (VIM-2) | ||
| None | 64 | 256 | 128 | 128 | 16 | 256 | |
| CP 35 |  | 64 | 128 | 32 | 32 | 16 | 256 |
| CP 56 |  | 32 | 256 | 32 | 64 | 8 | 256 |
| CP 57 |  | 32 | 256 | 32 | 32 | 4 | 256 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bersani, M.; Failla, M.; Vascon, F.; Gianquinto, E.; Bertarini, L.; Baroni, M.; Cruciani, G.; Verdirosa, F.; Sannio, F.; Docquier, J.-D.; et al. Structure-Based Optimization of 1,2,4-Triazole-3-Thione Derivatives: Improving Inhibition of NDM-/VIM-Type Metallo-β-Lactamases and Synergistic Activity on Resistant Bacteria. Pharmaceuticals 2023, 16, 1682. https://0-doi-org.brum.beds.ac.uk/10.3390/ph16121682
Bersani M, Failla M, Vascon F, Gianquinto E, Bertarini L, Baroni M, Cruciani G, Verdirosa F, Sannio F, Docquier J-D, et al. Structure-Based Optimization of 1,2,4-Triazole-3-Thione Derivatives: Improving Inhibition of NDM-/VIM-Type Metallo-β-Lactamases and Synergistic Activity on Resistant Bacteria. Pharmaceuticals. 2023; 16(12):1682. https://0-doi-org.brum.beds.ac.uk/10.3390/ph16121682
Chicago/Turabian StyleBersani, Matteo, Mariacristina Failla, Filippo Vascon, Eleonora Gianquinto, Laura Bertarini, Massimo Baroni, Gabriele Cruciani, Federica Verdirosa, Filomena Sannio, Jean-Denis Docquier, and et al. 2023. "Structure-Based Optimization of 1,2,4-Triazole-3-Thione Derivatives: Improving Inhibition of NDM-/VIM-Type Metallo-β-Lactamases and Synergistic Activity on Resistant Bacteria" Pharmaceuticals 16, no. 12: 1682. https://0-doi-org.brum.beds.ac.uk/10.3390/ph16121682