Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21WAF1/CIP1 and p27KIP1 in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Drug Treatment
2.3. Cell Viability Assessment Using MTT and Trypan Blue Dye Exclusion Method
2.4. Protein Preparation and Western Blot Analysis
2.5. Fluorescence Imaging for Cell Death Assessment
2.6. Statistical Analysis
3. Results
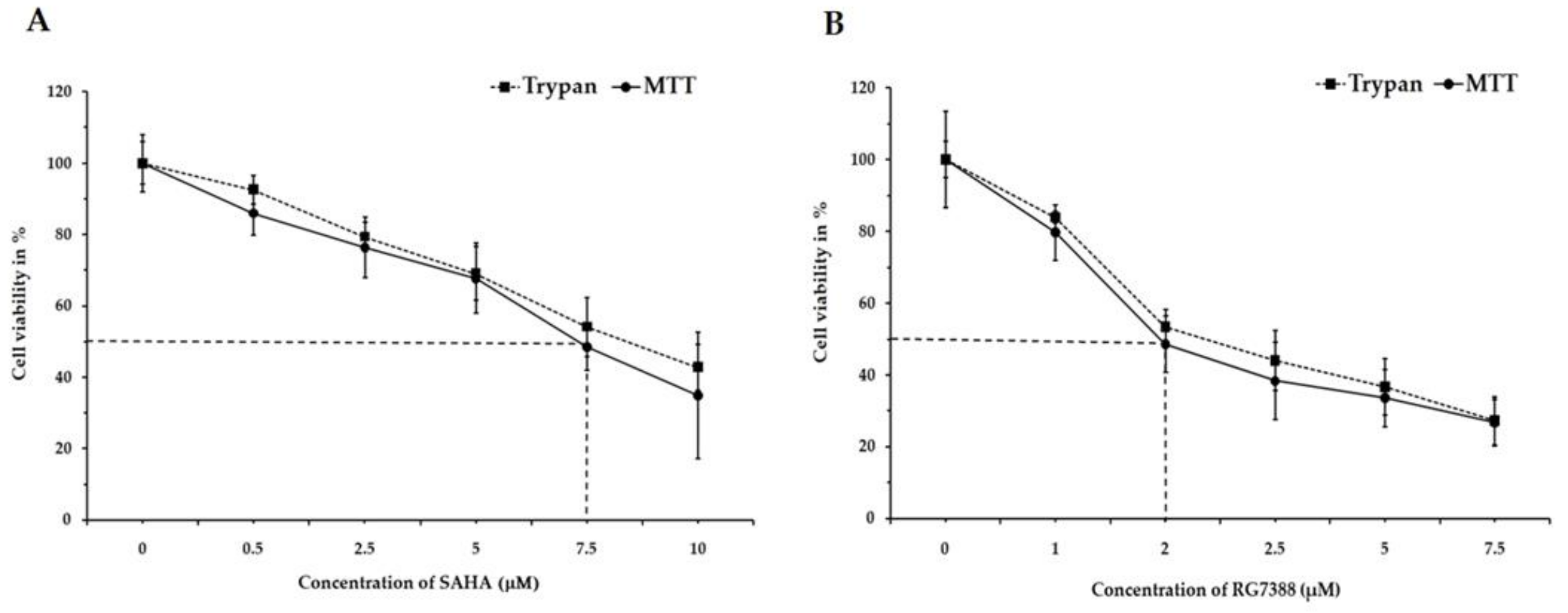
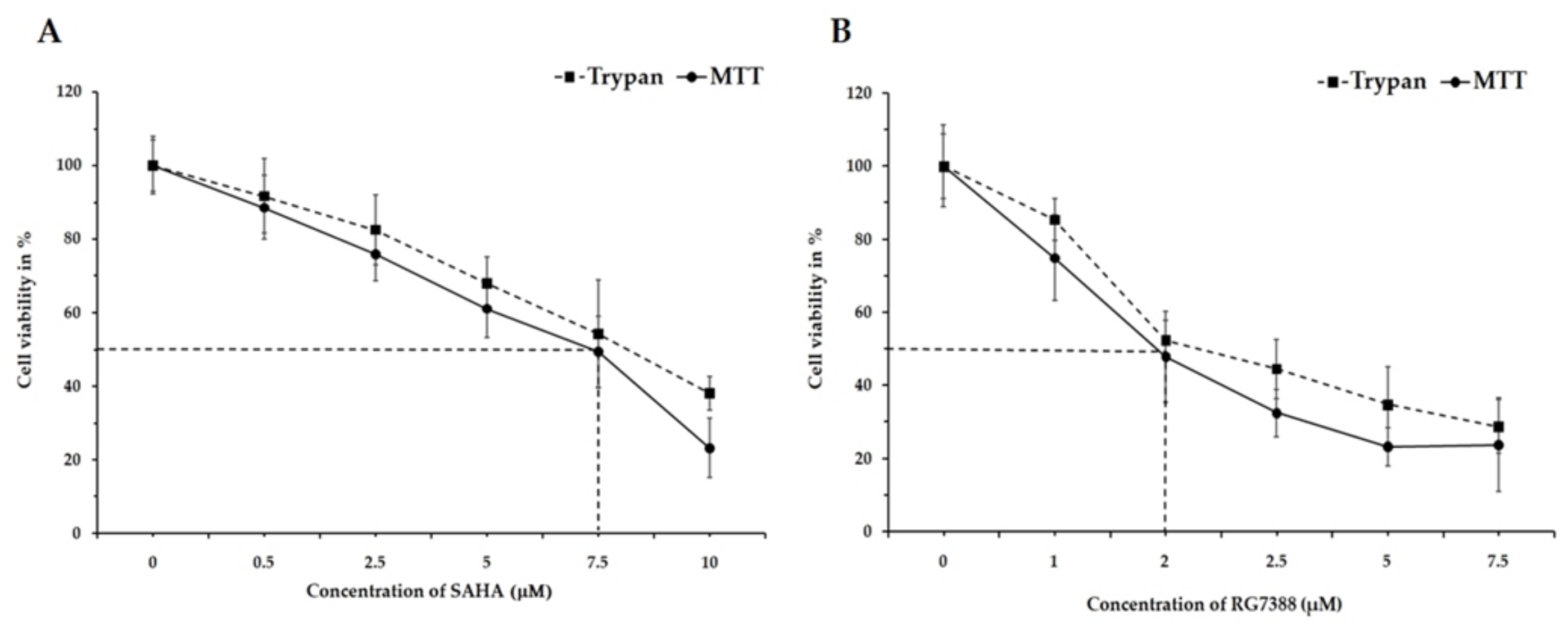
3.1. Changes in Cell Viability by SAHA and RG7388 Treatments in MCF-7 and LNCaP Cells
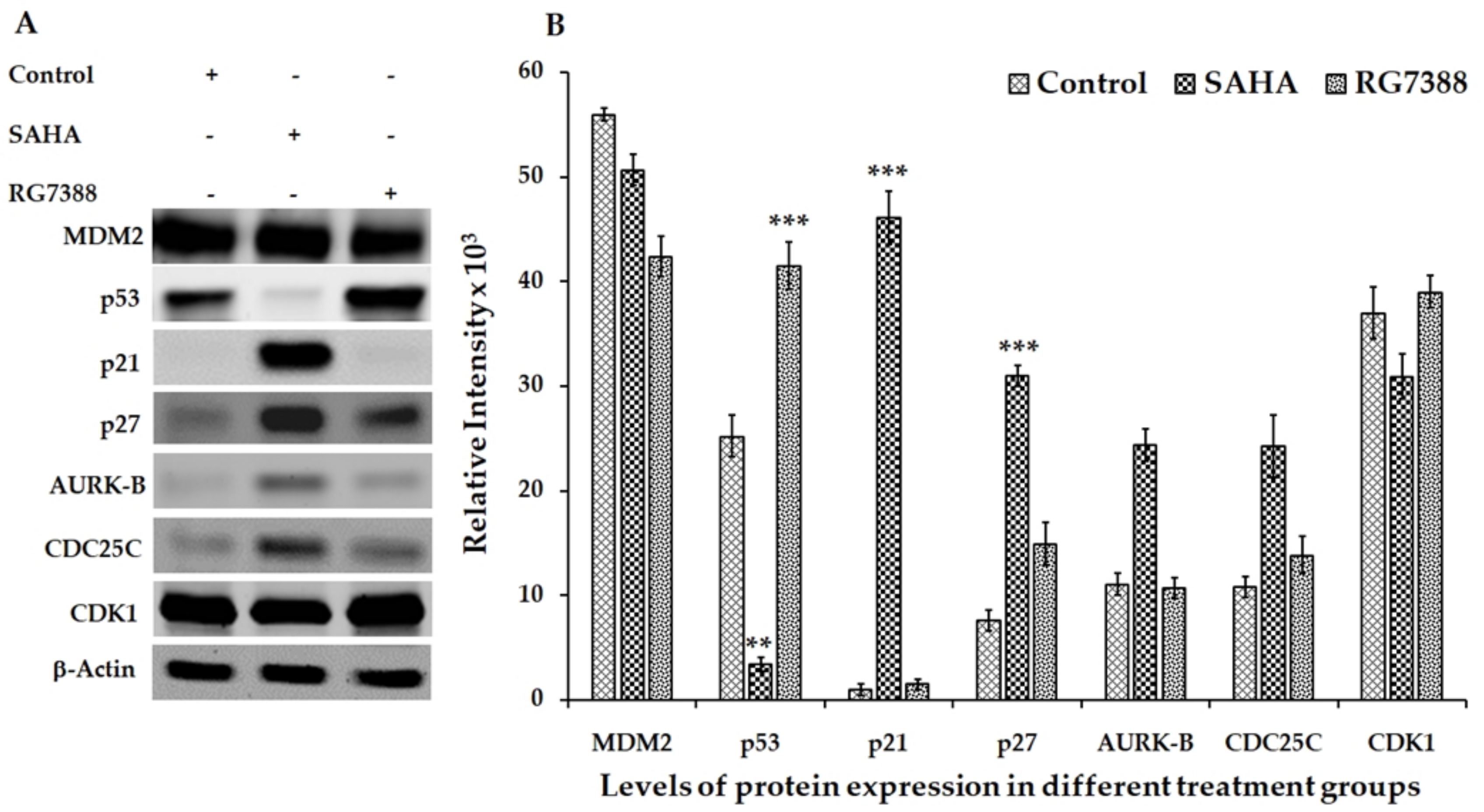
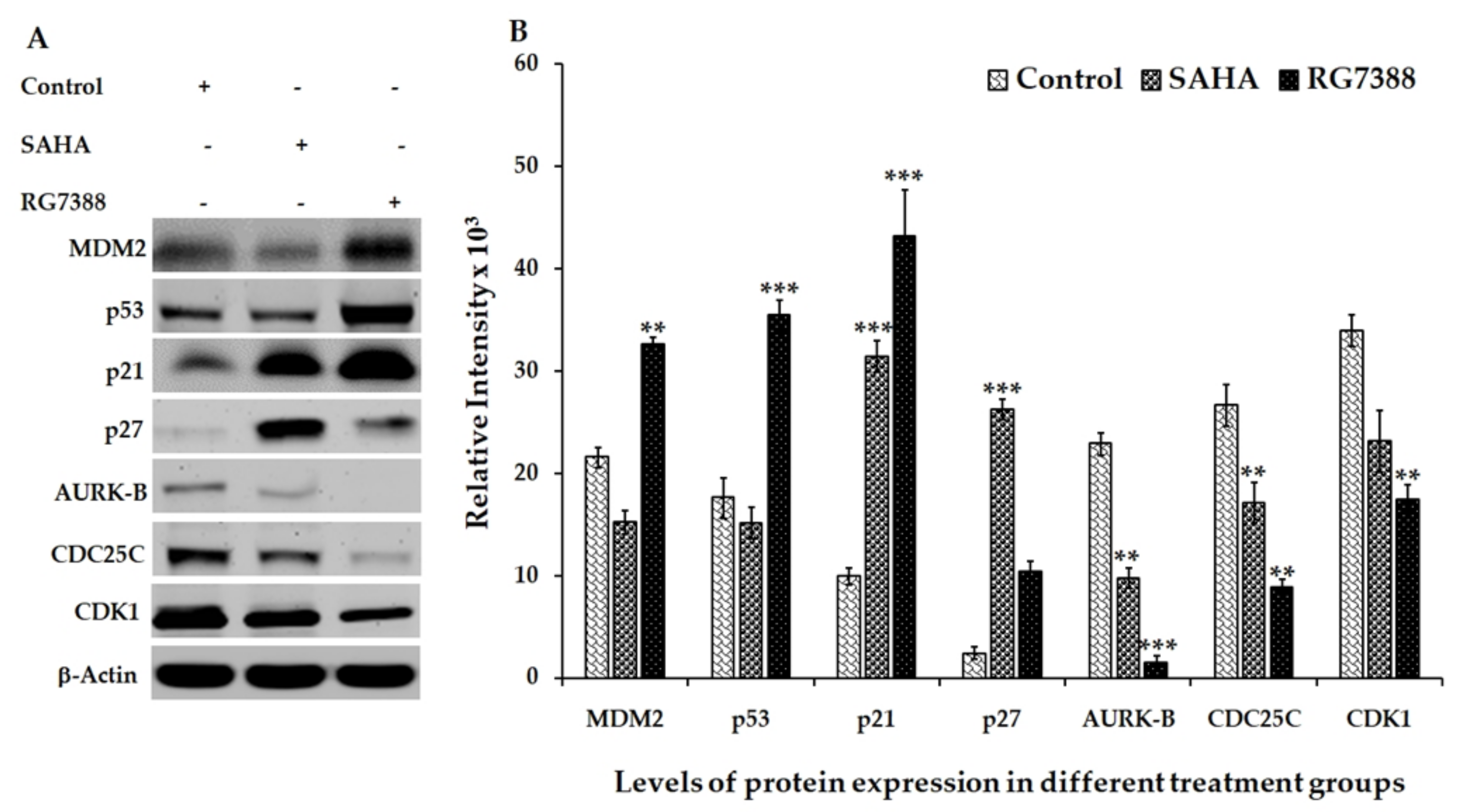
3.2. Effect of SAHA and RG7388 Treatments on p21WAF1/CIP1 and p27Kip1 Levels
3.3. Aurora Kinase-B (AURK-B), CDC25C, and CDK1 Levels in SAHA and RG7388 Treated Cells
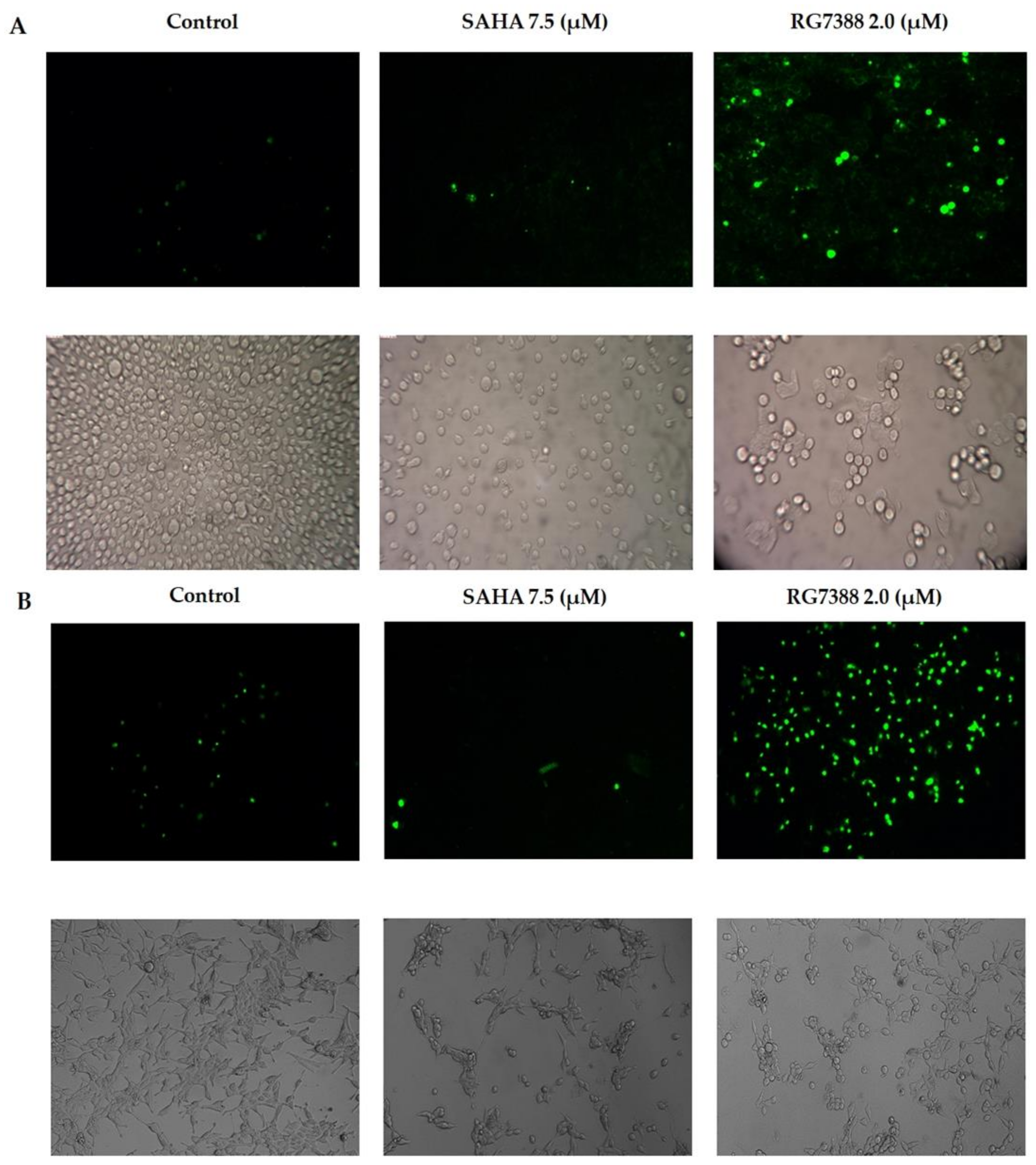
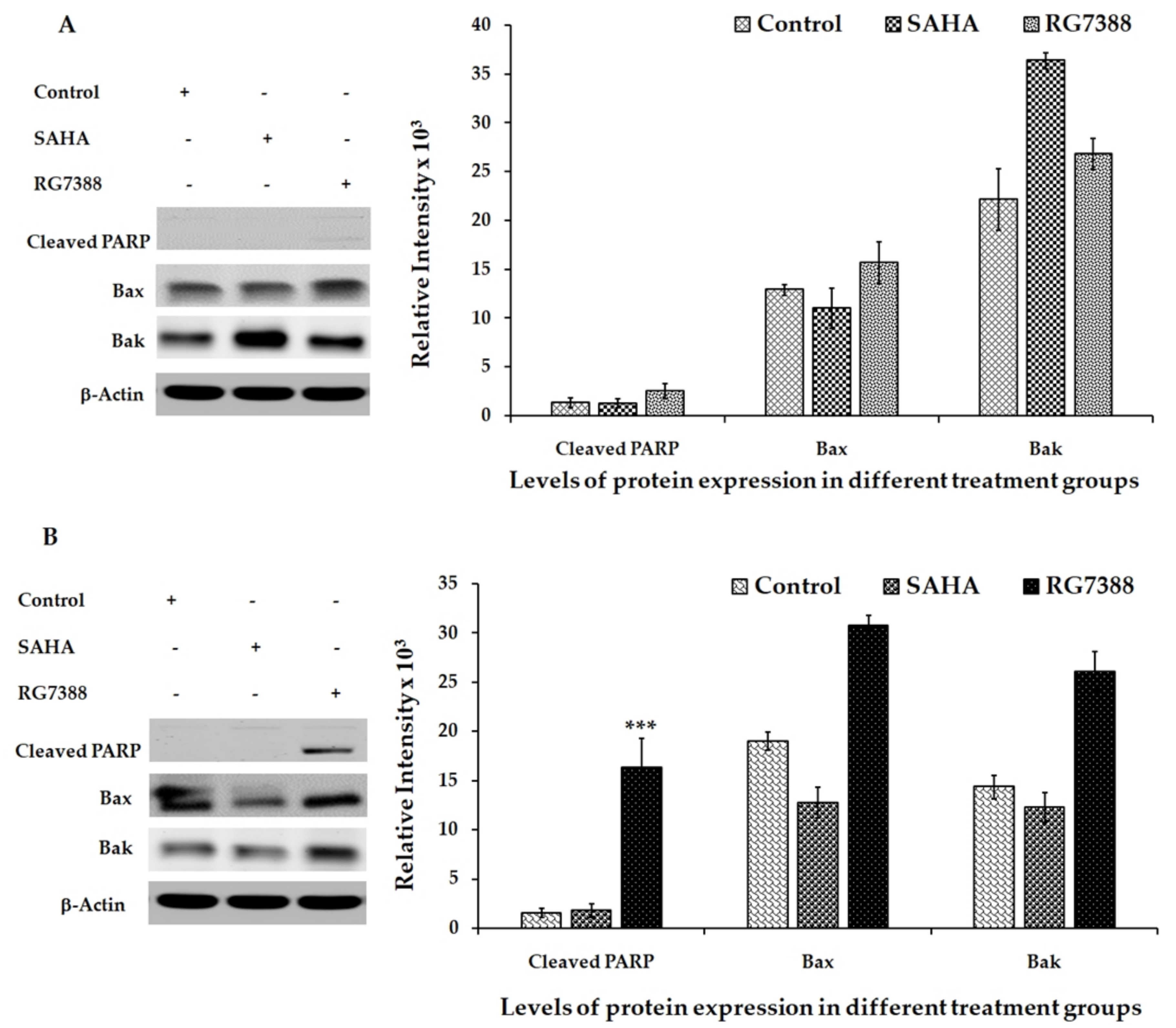
3.4. Apoptotic Effects of SAHA and RG7388 Treatments on MCF-7 and LNCaP Cells
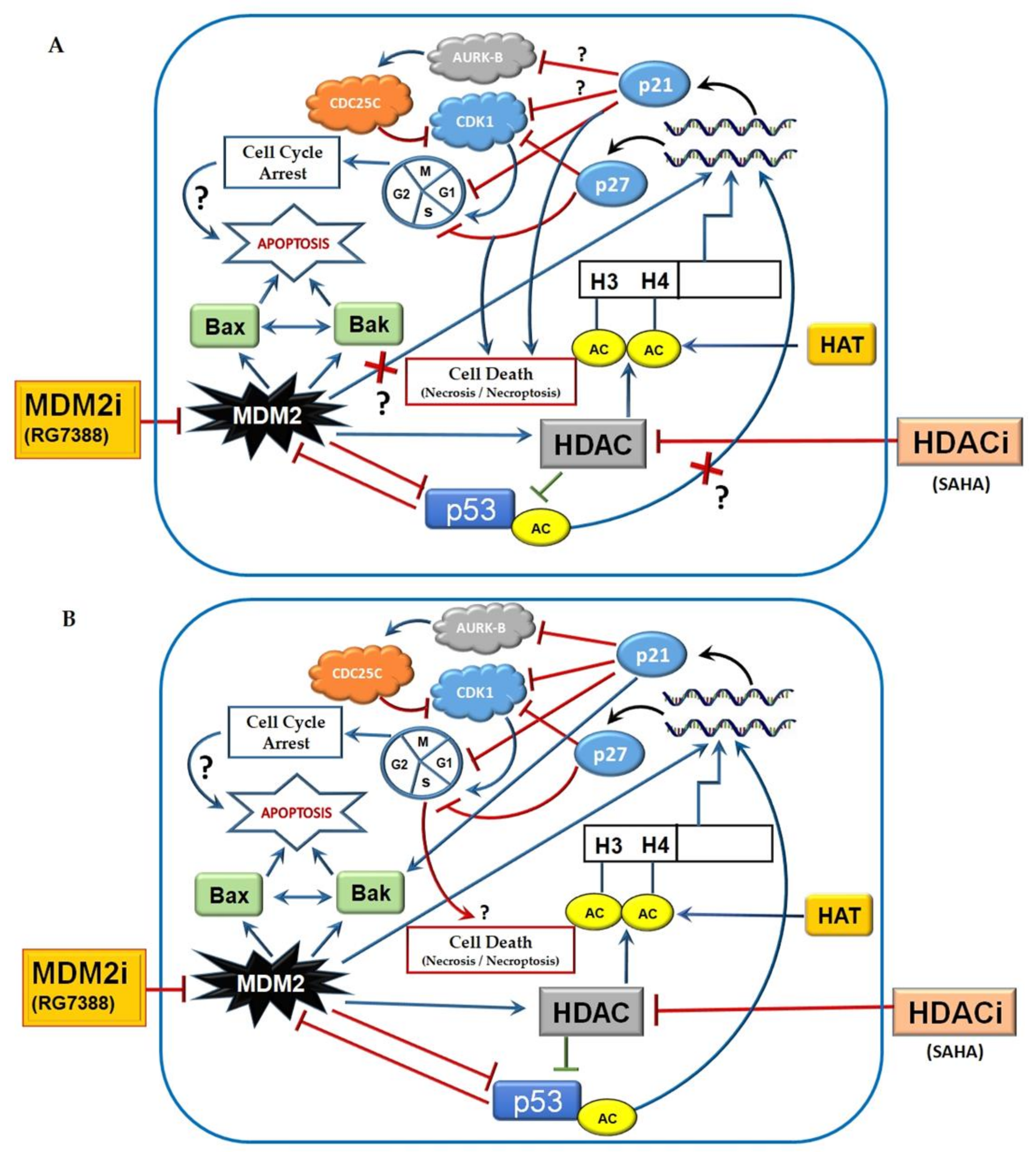
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cheng, M.H.; Wong, Y.H.; Chang, C.M.; Yang, C.C.; Chen, S.H.; Yuan, C.L.; Kuo, H.M.; Yang, C.Y.; Chiu, H.F. B1, a novel HDAC inhibitor, induces apoptosis through the regulation of STAT3 and NF-κB. Int. J. Mol. Med. 2017, 39, 1137–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Somasundar, P.; Yu, A.K.; Vona-Davis, L.; McFadden, D.W. Differential effects of Leptin on cancer in vitro. J. Surg. Res. 2003, 113, 50–55. [Google Scholar] [CrossRef]
- Connolly, R.; Stearns, V. Epigenetics as a therapeutic target in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Troso-Sandoval, T.; Rosen, N.; Rifkind, R.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor suberoylanilidehydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001, 61, 8492–8497. [Google Scholar] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef]
- Weiqiang, Z.; Xiuyan, F.; Han, H.; Shanchun, G.; Guangdi, W. Synergistic effects of combined treatment with histone deacetylase inhibitor suberoylanilidehydroxamic acid and TRAIL on human breast cancer cells. Sci. Rep. 2016, 6, 28004. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Brit. J. Cancer. 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [Green Version]
- Lidia, C.S.; Denise, D.F.; Sonya, Z.; Paul, O.K.; Helena, C.; Nancy, T.; Hong, X.; Dalia, C. Histone Deacetylase Inhibition Selectively Alters the Activity and Expression of Cell Cycle Proteins Leading to Specific Chromatin Acetylation and Antiproliferative Effects. J. Biol. Chem. 1999, 274, 34940–34947. [Google Scholar] [CrossRef] [Green Version]
- Tomas, E.; Johana, P.; Marie, S.; Jan, H. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Rathinavelu, A.; Narasimhan, M.; Muthumani, P. A Novel Regulation of VEGF Expression by HIF-1α and STAT3 in HDM2 transfected Prostate Cancer Cells. J. Cell. Mol. Med. 2011, 16, 1472. [Google Scholar] [CrossRef] [PubMed]
- Fakharzadeh, S.S.; Trusko, S.P.; George, D.L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991, 10, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Gilkes, D.M.; Farooqi, B.; Sebti, S.M.; Chen, J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J. Biol. Chem. 2006, 281, 33030–33035. [Google Scholar] [CrossRef]
- Sigalas, I.; Calvert, A.H.; Anderson, J.J.; Neal, D.E.; Lunec, J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: Transforming ability and frequent detection in human cancer. Nat. Med. 1996, 2, 912–917. [Google Scholar] [CrossRef]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [Green Version]
- Bartel, F.; Pinkert, D.; Fiedler, W.; Kappler, M.; Wurl, P.; Schmidt, H.; Taubert, H. Expression of alternative and aberrantly spliced transcripts of the MDM2 mRNA is not tumor-specific. Int. J. Oncol. 2004, 24, 143–151. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Zell, J.A.; Malave, A.; Rathinavelu, A. Expression of Vascular Endothelial Growth Factor mRNA in GI-101A and HL-60 Cell Lines. Bioche. Biophys. Res. Commun. 2000, 270, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Rose, R.; Karthikeyan, M.; Rathinavelu, A. Detection of HDM2 and VEGF co-expression in cancer cell lines: Novel effect of HDM2 antisense treatment on VEGF expression. Life Sci. 2007. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Lakoma, A.; Barbieri, E.; Agarwal, S.; Jackson, J.; Chen, Z.; Kim, Y.; McVay, M.; Shohet, J.M.; Kim, E.S. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015, 15026. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.T.; Vassilev, L. Small-molecule inhibitors of the p53-MDM2 interaction. Curr. Top. Microbiol. Immunol. 2011, 348, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Katayose, Y.; Kim, M.; Rakkar, A.N.; Li, Z.; Cowan, K.H.; Seth, P. Promoting apoptosis: A novel activity associated with the cyclin-dependent kinase inhibitor p27. Cancer Res. 1997, 57, 5441–5445. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef]
- Polyak, K.; Lee, M.H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massagu, Ã.J. Cloning of p27Klpl, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Lili, H.; Yoshihiro, S.; Toshiyuki, S.; Arthur, B.P. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilidehydroxamic acid (SAHA) through the Sp1 sites. Oncogene 2000, 19, 5712–5719. [Google Scholar] [CrossRef]
- Purva, B.; Michael, P.; Ramona, S.; Warren, F.; Hirohito, Y.; Maria, B.; Kathy, R.; Hong-Gang, W.; Victoria, R.; Kapil, B. Activity of SuberoylanilideHydroxamic Acid Against Human Breast Cancer Cells with Amplification of Her-2. Clin. Cancer Res. 2005, 11, 6382. [Google Scholar] [CrossRef]
- Cohen, L.A.; Marks, P.A.; Rifkind, R.A.; Amin, S.; Desai, D.; Pittman, B.; Richon, V.M. Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen induced mammary tumors. Anticancer Res. 2002, 22, 1497–1504. [Google Scholar] [PubMed]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice lacking p27(Kipl) display increased body size, multiple organ hyperplasia, retinal dysplasia and pituitary tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kipl)-deficient mice. Cell 1996, 85, 733–744. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Kineman, R.D.; Manova-Todorova, K.O.; Scares, V.C.; Hoffman, E.S.; Ono, M.; Khanam, D.; Mayday, A.C.; Frohman, L.A.; Koff, A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kipl). Cell 1996, 85, 721–732. [Google Scholar] [CrossRef]
- Craig, C.; Wersto, R.; Kim, M.; Ohri, E.; Li, Z.; Katayose, D.; Lee, S.J.; Trepel, J.; Cowan, K.; Seth, P. A recombinant adenovirus expressing p27Kipl induces cell cycle arrest and loss of cyclin-Cdk activity in human breast cancer cells. Oncogene 1997, 14, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.M.; Victoria, M.R.; Thomas, M.; William, K.K. Histone Deacetylase Inhibitors. Adv. Cancer Res. 2004, 91, 137–168. [Google Scholar]
- Amila, S.; Kenneth, J.O.; Derek, J.R. Combination Therapy with Histone Deacetylase inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 29, 92. [Google Scholar] [CrossRef]
- Abigail, B.M.L.; James, P.S.; Suzanne, E.W.; Holly, M.A.; Ching-yi, C.; Donald, P.M. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2017, 78, 266–277. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men with Metastatic Castration-Resistant Prostate Cancer Treated with First-and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Umamaheswari, N.; Thiagarajan, V.; Vijayaraghavan, R.; Shila, S.; Rathinavelu, A. Comparative Effects of HDAC Inhibitor SAHA and MDM2 Inhibitor RG7388 in LNCaP Prostate Cancer Cells. Biomed. J. Sci. Tech. Res. 2018, 8. [Google Scholar] [CrossRef]
- Felix, Y.F.; Yu, Z.; Vishal, K.; Joseph, R.E.; William, C.J.; Wei, C.; Skyler, B.J.; Connor, L.; Shaomeng, W.; Daniel, A.H. Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a p53-Dependent Manner. Neoplasia 2016, 18, 213–222. [Google Scholar] [CrossRef]
- A trial of idasanutlin with abiraterone or enzalutamide for men with prostate cancer who haven’t had docetaxel (MAdCaP). Cancer Research UK. Available online: https://www.cancerresearchuk.org/about-cancer/find-a-clinical-trial/a-trial-of-idasanutlin-with-abiraterone-or-enzalutamide-for-men-with-prostate-cancer-who-havent-had#undefined (accessed on 17 January 2019).
- Schwarze, S.R.; Shi, Y.; Fu, V.X.; Watson, P.A.; Jarrard, D.F. Role of cyclin-dependent kinase inhibitors in the growth arrest at senescence in human prostate epithelial and uroepithelial cells. Oncogene 2001, 20, 8184–8192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanesa, D.M.; Choudhury, M.S.; Mallouh, C.; Tazaki, H.; Konno, S. Methylglyoxal-induced apoptosis in human prostate carcinoma: Potential modality for prostate cancer treatment. Eur. Urol. 2000, 37, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Tsihlias, J.; Zhang, W.; Bhattacharya, N.; Flanagan, M.; Klotz, L.; Slingerland, J. Involvement of p27Kip1 in G1 arrest by high dose 5-dihydrotestosterone in LNCaP human prostate cancer cells. Oncogene 2000, 19, 670–679. [Google Scholar] [CrossRef]
- Singh, G.; Lakkis, C.L.; Laucirica, R.; Epner, D.E. Regulation of prostate cancer cell division by glucose. J. Cell. Physiol. 1999, 180, 431–438. [Google Scholar] [CrossRef]
- Aaltomaa, S.; Eskelinen, M.; Lipponen, P. Expression of cyclin A and D proteins in prostate cancer and their relation to clinicopathological variables and patient survival. Prostate 1999, 38, 175–182. [Google Scholar] [CrossRef]
- Guinan, P.; Shaw, M.; Mirochnik, Y.; Slobodskoy, L.; Ray, V.; Rubenstein, M. Paclitaxel is more effective than thalidomide in inhibiting LNCaP tumor growth in a prostate cancer model. Methods Findings. Exp. Clin. Pharmacol. 1998, 20, 739–742. [Google Scholar]
- Perry, J.E.; Grossmann, M.E.; Tindall, D.J. Epidermal growth factor induces cyclin D1 in a human prostate cancer cell line. Prostate 1998, 35, 117–124. [Google Scholar] [CrossRef]
- Itoh, N.; Kakehi, Y.; Akao, T.; Kinoshita, H.; Okada, Y.; Yoshida, O. Concomitant presence of p16/cyclin-dependent kinase 4 and cyclin D/cyclin-dependent kinase 4 complexes in LNCaP prostatic cancer cell line. Jpn. J. Cancer Res. 1997, 88, 229–233. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Bartkova, J.; Lukas, J. The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 1996, 8, 805–814. [Google Scholar] [CrossRef]
- Kato, J.; Matsushime, H.; Heibert, S.W.; Ewen, M.E.; Sherr, C.J. Direct binding of cyclin D to the retinoblastoma gene product(pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar] [PubMed]
- Furuya, Y.; Akimoto, S.; Yasuda, K.; Ito, H. Apoptosis of androgen-independent prostate cell line induced by inhibition of fatty acid synthesis. Anticancer Res. 1997, 17, 4589–4593. [Google Scholar] [PubMed]
- Naveen, K.; Vincent, J.G. The role of treatment modality on the utility of predictive tissuebiomarkers in clinical prostate cancer: A systematic review. J. Cancer Res. Clin. Oncol. 2013, 139, 1–24. [Google Scholar] [CrossRef]
- Vis, A.N.; Noordzij, M.A.; Fitoz, K.; Wildhagen, M.F.; Schroder, F.H.; van der Kwast, T.H. Prognostic value of cell cycle proteinsp27(kip1) and MIB-1, and the cell adhesion protein CD44 insurgically treated patients with prostate cancer. J. Urol. 2000, 164, 2156–2161. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rasappan, P.; Rathinavelu, A. Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21WAF1/CIP1 and p27KIP1 in Cancer Cells. Medicina 2019, 55, 30. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020030
Natarajan U, Venkatesan T, Radhakrishnan V, Samuel S, Rasappan P, Rathinavelu A. Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21WAF1/CIP1 and p27KIP1 in Cancer Cells. Medicina. 2019; 55(2):30. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020030
Chicago/Turabian StyleNatarajan, Umamaheswari, Thiagarajan Venkatesan, Vijayaraghavan Radhakrishnan, Shila Samuel, Periannan Rasappan, and Appu Rathinavelu. 2019. "Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21WAF1/CIP1 and p27KIP1 in Cancer Cells" Medicina 55, no. 2: 30. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020030