Impact of Hydrous Manganese and Ferric Oxides on the Behavior of Aqueous Rare Earth Elements (REE): Evidence from a Modeling Approach and Implication for the Sink of REE



Abstract
:1. Introduction
2. Materials and Methods
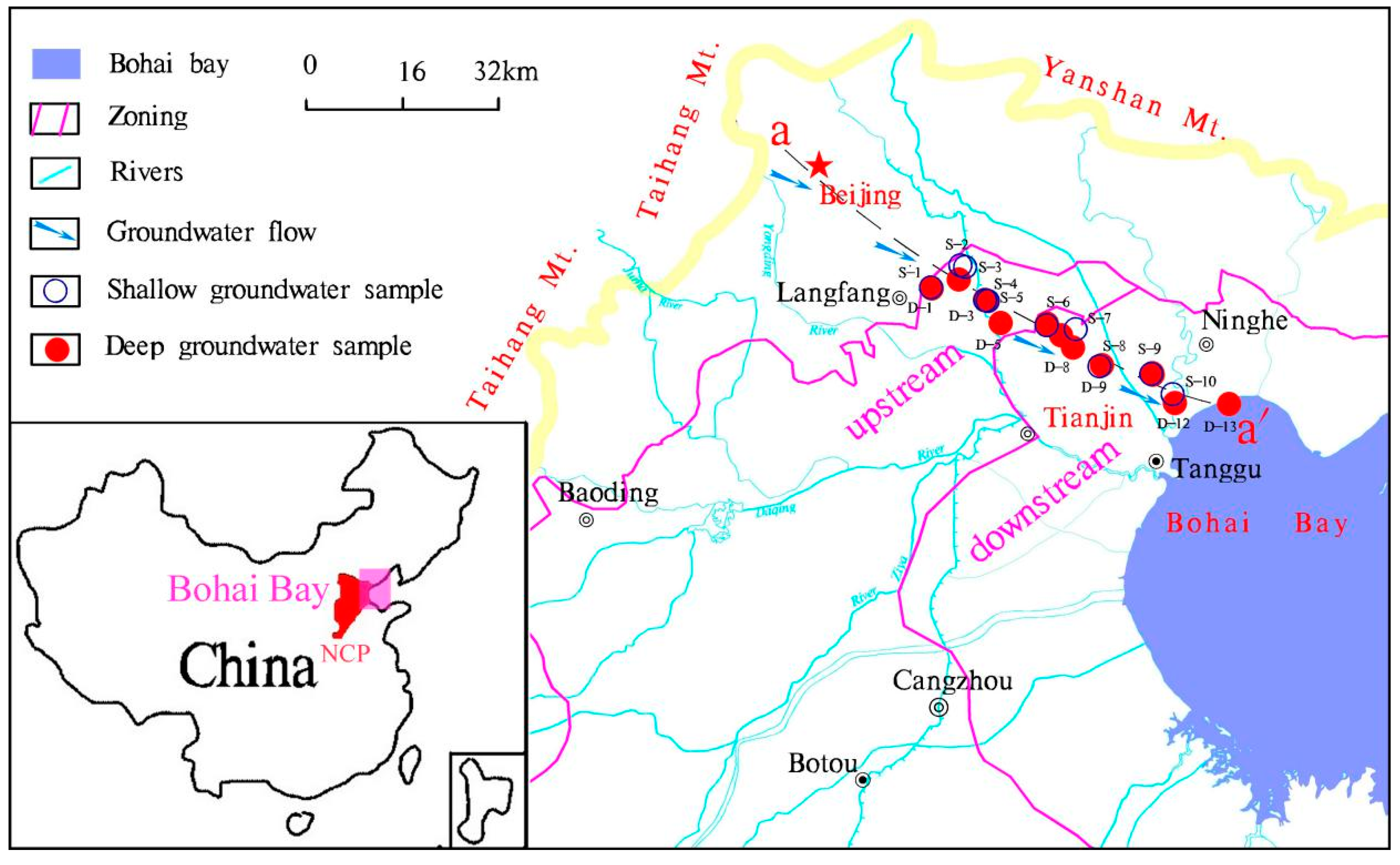
2.1. Regional Hydrogeological Settings
2.2. Groundwater Sampling
2.3. Chemical Analysis
2.4. Surface Complexation Modeling
3. Results
3.1. General Groundwater Chemistry
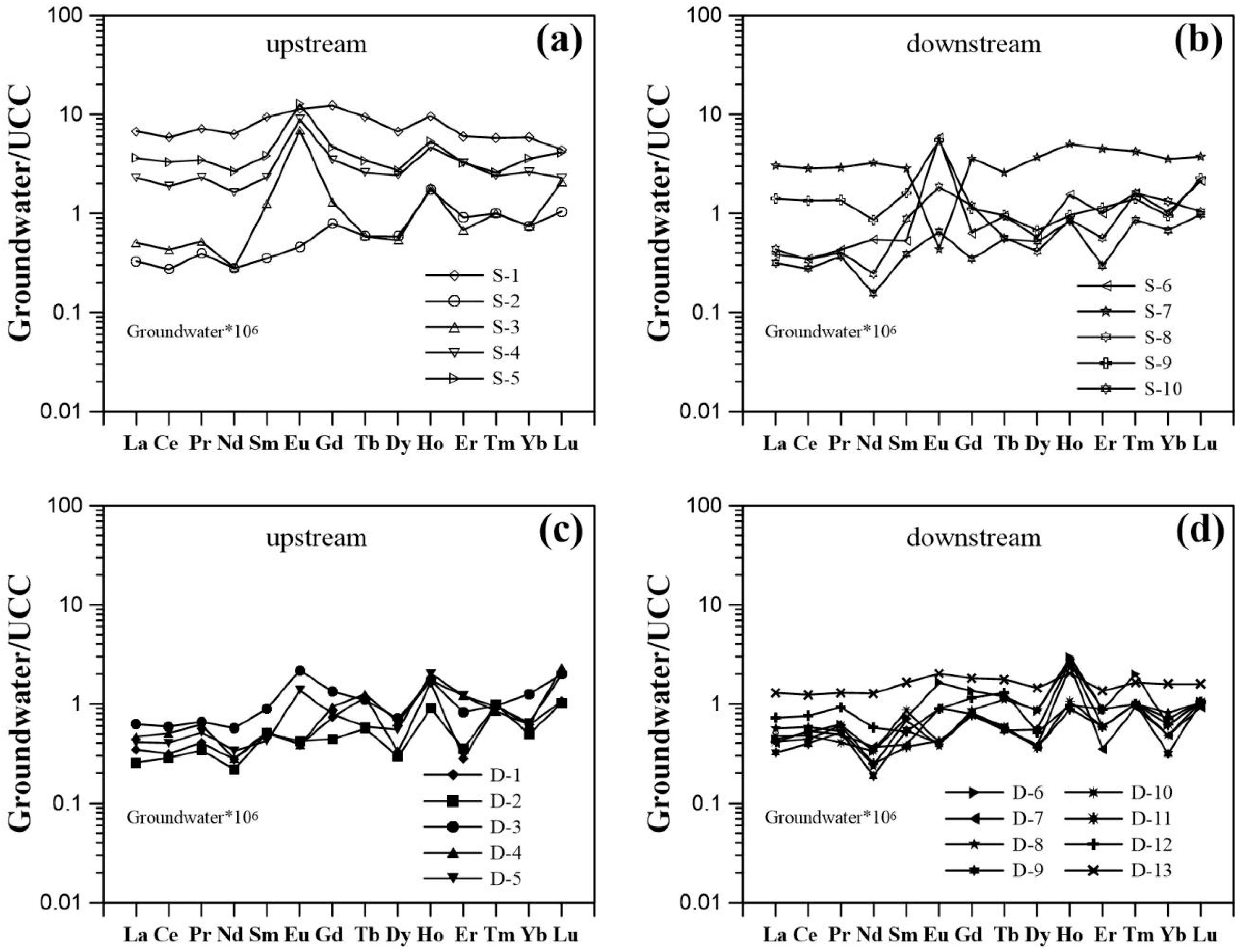
3.2. REE Concentrations and Normalized Patterns
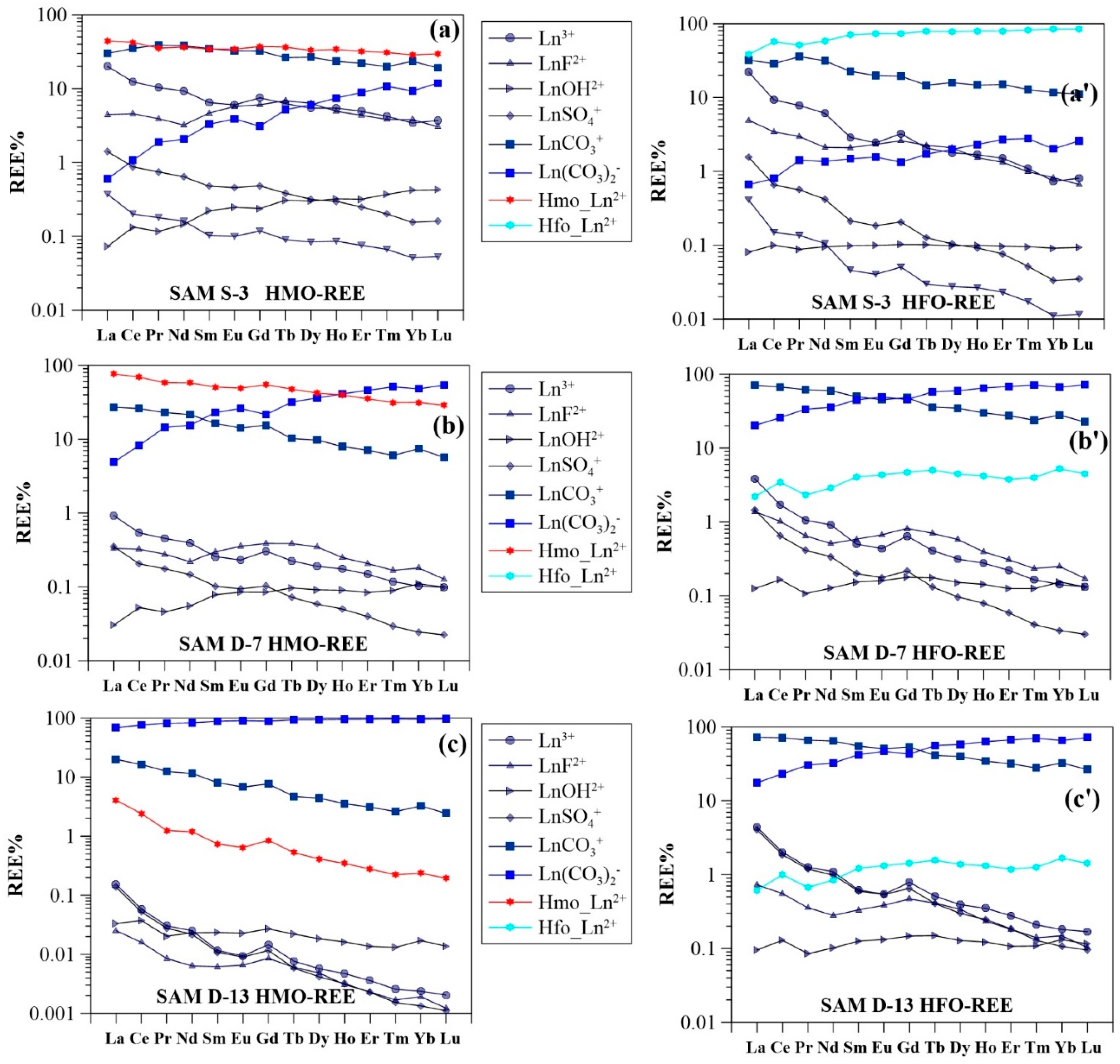
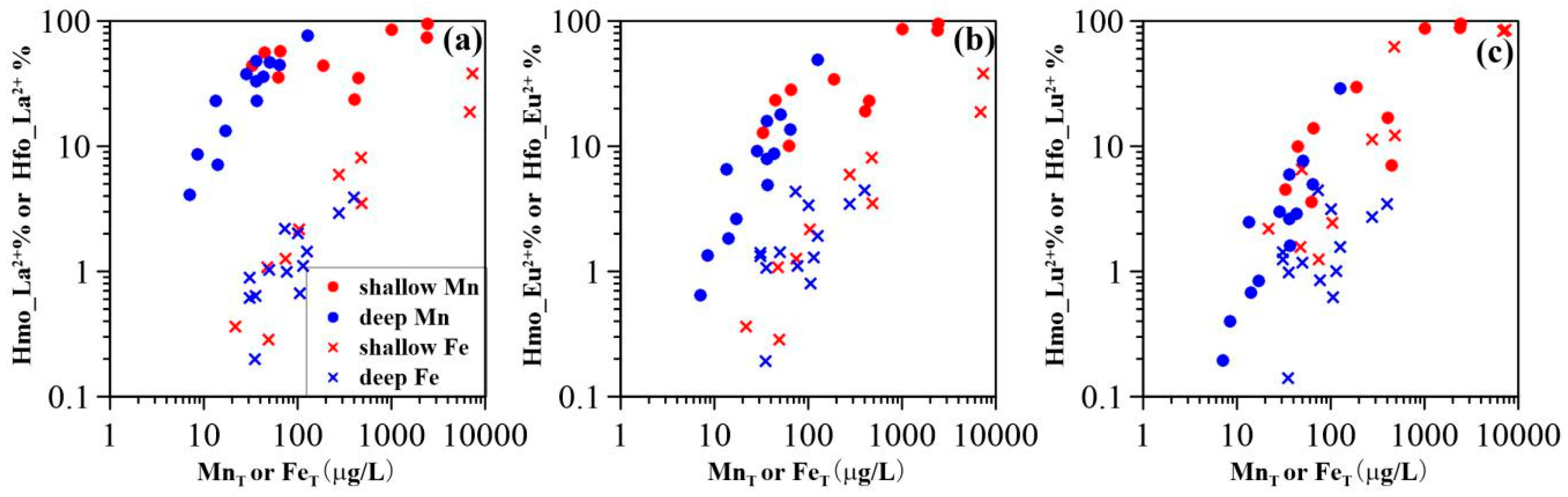
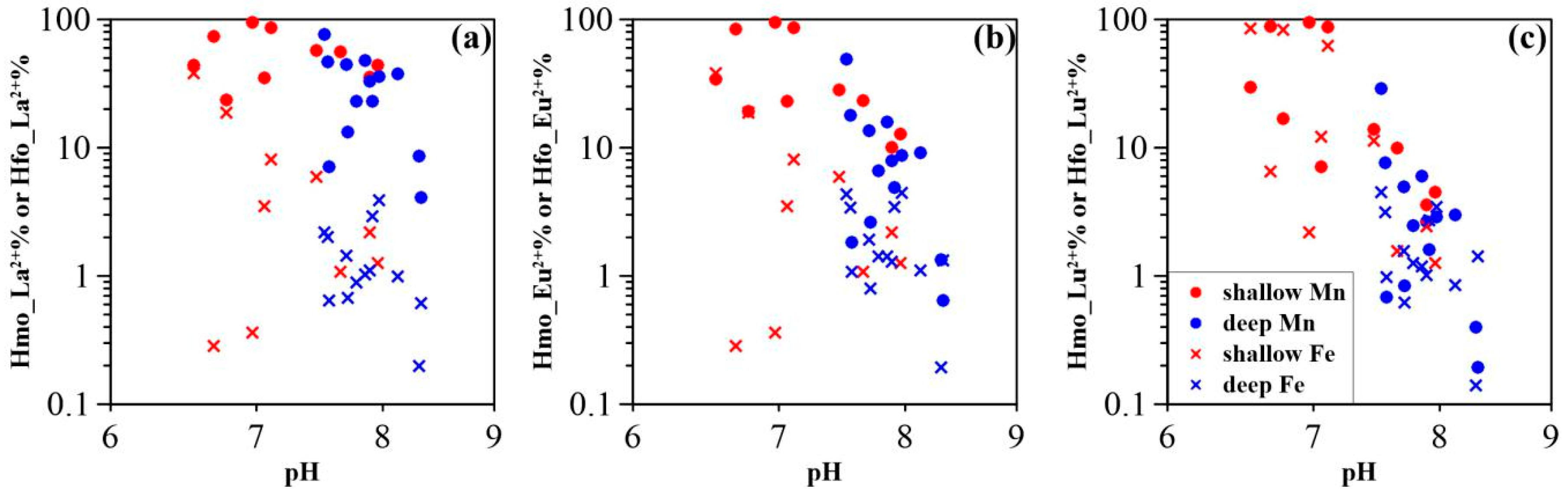
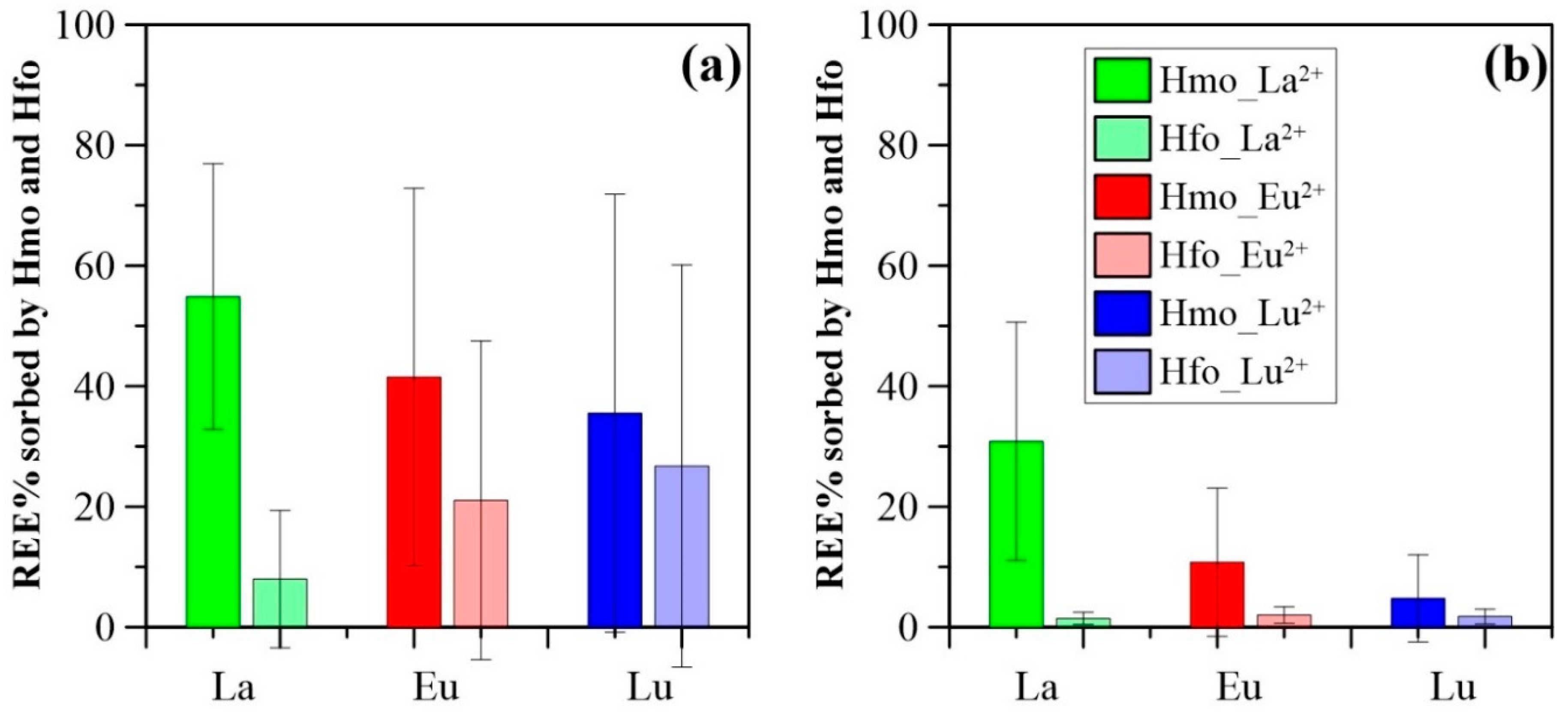
3.3. Surface Complexation Modeling Results
4. Discussion
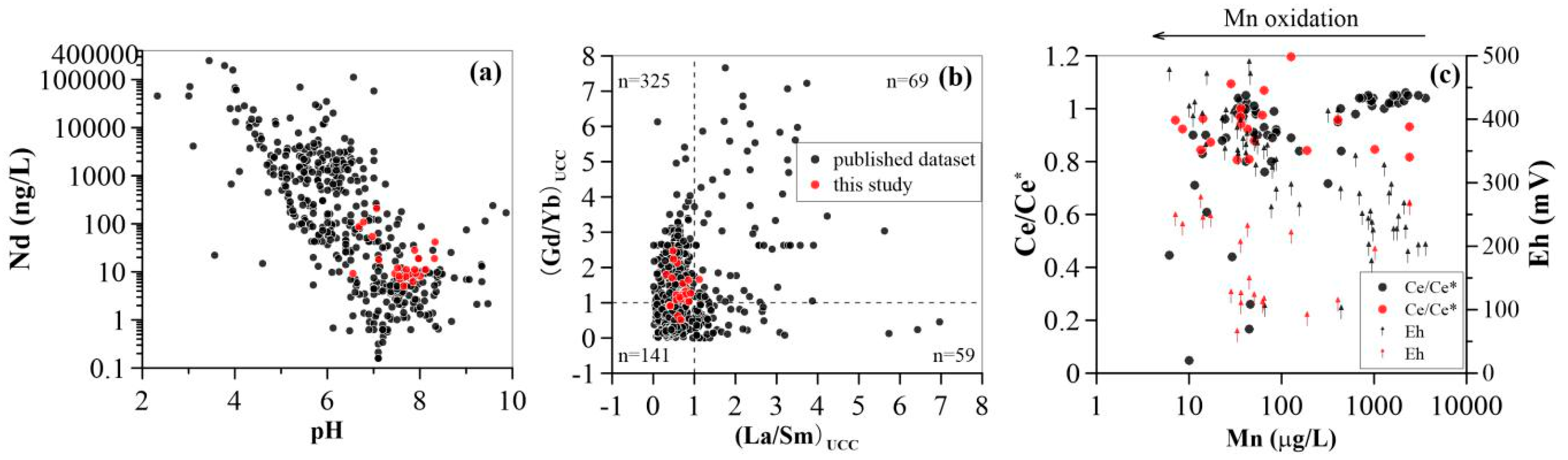
4.1. Mobility of REE in Groundwater
4.2. Impact of Manganese
4.3. Impact of Iron
4.4. Comparison of HMO Scavenging Model and HFO Scavenging Model
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Sample No. | Distance (km) | Depth (m) | Eh (mV) | pH | Conductivity (μs/cm) | TDS (mg/L) | F− (mg/L) | Cl− (mg/L) | NO3− (mg/L) | SO42− (mg/L) | HCO3− (mg/L) | K+ (mg/L) | Na+ (mg/L) | Ca2+ (mg/L) | Mg2+ (mg/L) | FeT (μg/L) | MnT (μg/L) | TOC (mg/L) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shallow | Upstream | S-1 | 81 | 12 | 96 | 7.1 | 4374 | 3260 | 5.2 | 977 | 127 | 557 | 783 | 1.7 | 751 | 183 | 268 | 485 | 444 | 2.2 |
| S-2 | 83 | 150 | 100 | 7.5 | 1094 | 850 | 2.1 | 43 | 15 | 97 | 648 | 0.8 | 310 | 31 | 26 | 276 | 65 | 2.7 | ||
| S-3 | 84 | 24 | 86 | 6.6 | 1452 | 968 | 1.7 | 200 | 4 | 5 | 749 | 2.2 | 118 | 194 | 68 | 7382 | 189 | 2.2 | ||
| S-4 | 96 | 7.3 | 261 | 7.0 | 3179 | 2576 | 3.7 | 801 | 138 | 311 | 677 | 2.9 | 486 | 356 | 139 | 122 | 2430 | 2.2 | ||
| S-5 | 97 | 6 | 262 | 6.7 | 3953 | 3343 | 2.0 | 961 | 414 | 454 | 703 | 0.3 | 554 | 437 | 169 | 249 | 2401 | 2.5 | ||
| S-6 | 112 | 150 | 189 | 7.1 | 7398 | 6250 | 12.2 | 2605 | 90 | 589 | 840 | 1.9 | 2009 | 226 | 296 | 476 | 1015 | 4.6 | ||
| Downstream | S-7 | 128 | 50 | 109 | 6.8 | 10050 | 9359 | 8.6 | 3036 | 134 | 1511 | 1611 | 13.8 | 3008 | 283 | 522 | 6912 | 407 | 1.5 | |
| S-8 | 140 | 175 | 60 | 8.0 | 511 | 418 | 2.7 | 31 | 9 | 22 | 291 | 0.4 | 182 | 9 | 2 | 75 | 33 | 1.5 | ||
| S-9 | 170 | 170 | 144 | 7.7 | 483 | 392 | 0.7 | 45 | 3 | 23 | 279 | 0.5 | 151 | 17 | 3 | 48 | 45 | 1.6 | ||
| S-10 | 179 | 160 | 106 | 7.9 | 569 | 460 | 0.8 | 75 | 2 | 21 | 287 | 0.5 | 185 | 22 | 5 | 104 | 62 | 1.8 | ||
| Deep | Upstream | D-1 | 75 | 320 | 271 | 7.8 | 596 | 439 | 2.1 | 46 | 8 | 71 | 317 | 0.5 | 178 | 13 | 3 | 31 | 13 | 0.9 |
| D-2 | 72 | 303 | 239 | 7.6 | 955 | 528 | 1.3 | 119 | 0 | 101 | 330 | 0.3 | 114 | 11 | 6 | 36 | 14 | 1.8 | ||
| D-3 | 66 | 390 | 225 | 8.0 | 783 | 606 | 1.1 | 68 | 7 | 29 | 405 | 0.8 | 254 | 20 | 7 | 397 | 43 | 3.0 | ||
| D-4 | 65 | 330 | 104 | 7.9 | 752 | 563 | 1.0 | 57 | 0 | 27 | 405 | 0.6 | 235 | 15 | 6 | 276 | 37 | 3.0 | ||
| D-5 | 62 | 400 | 241 | 7.7 | 989 | 621 | 1.4 | 55 | 4 | 5 | 573 | 0.6 | 221 | 10 | 4 | 106 | 17 | 2.3 | ||
| Downstream | D-6 | 56 | 375 | 120 | 7.9 | 707 | 551 | 3.7 | 33 | 3 | 21 | 429 | 0.6 | 246 | 11 | 3 | 115 | 36 | 2.8 | |
| D-7 | 54 | 400 | 215 | 7.5 | 1280 | 929 | 1.1 | 224 | 4 | 33 | 537 | 0.7 | 326 | 46 | 24 | 73 | 127 | 2.9 | ||
| D-8 | 50 | 260 | 111 | 7.7 | 617 | 533 | 3.9 | 34 | 11 | 12 | 462 | 0.7 | 208 | 19 | 6 | 126 | 65 | 2.4 | ||
| D-9 | 42 | 235 | 200 | 7.9 | 510 | 396 | 2.2 | 39 | 6 | 27 | 258 | 0.4 | 163 | 11 | 2 | 50 | 36 | 1.3 | ||
| D-10 | 22 | 300 | 121 | 8.1 | 427 | 347 | 1.1 | 20 | 6 | 10 | 311 | 0.4 | 127 | 11 | 3 | 77 | 28 | 2.4 | ||
| D-11 | 21 | 240 | 118 | 7.6 | 493 | 414 | 1.8 | 39 | 2 | 16 | 323 | 1.2 | 169 | 14 | 9 | 101 | 51 | 2.1 | ||
| D-12 | 6 | 500 | 228 | 8.3 | 694 | 548 | 3.2 | 43 | 3 | 19 | 359 | 0.5 | 265 | 6 | 1 | 35 | 9 | 2.8 | ||
| D-13 | 0 | 600 | 243 | 8.3 | 585 | 506 | 1.3 | 37 | 7 | 72 | 258 | 0.4 | 228 | 6 | 1 | 31 | 7 | 2.2 | ||
| Location | Sample No. | La | Ce | Pr | Nd | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | ∑REE | Eu/Eu* | Ce/Ce* | (Gd/Nd)ucc | (Yb/Nd)ucc | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shallow | Upstream | S-1 | 216.1 | 426.9 | 56.9 | 208.9 | 53.2 | 14.1 | 63.9 | 8.0 | 38.8 | 10.0 | 20.5 | 2.9 | 18.2 | 2.1 | 1141 | 1.1 | 0.8 | 1.9 | 0.9 |
| S-2 | 10.5 | 19.9 | 3.1 | 9.2 | 2.0 | 0.6 | 4.1 | 0.5 | 3.4 | 1.8 | 3.1 | 0.5 | 2.3 | 0.5 | 61 | 0.9 | 0.8 | 2.8 | 2.7 | ||
| S-3 | 16.1 | 31.4 | 4.1 | 9.3 | 7.2 | 8.6 | 6.8 | 0.5 | 3.1 | 1.9 | 2.3 | 0.5 | 2.3 | 1.0 | 95 | 5.4 | 0.8 | 4.6 | 2.6 | ||
| S-4 | 73.3 | 137.4 | 18.3 | 54.0 | 13.2 | 11.1 | 18.1 | 2.2 | 14.1 | 4.8 | 11.1 | 1.2 | 8.2 | 1.1 | 368 | 3.1 | 0.8 | 2.1 | 1.6 | ||
| S-5 | 116.2 | 241.0 | 27.4 | 88.0 | 21.9 | 15.7 | 24.1 | 2.9 | 15.9 | 5.6 | 11.0 | 1.3 | 11.1 | 2.0 | 584 | 3.0 | 0.9 | 1.7 | 1.3 | ||
| S-6 | 12.4 | 25.2 | 3.4 | 18.0 | 3.0 | 7.2 | 3.3 | 0.8 | 3.3 | 1.6 | 3.4 | 0.8 | 3.2 | 1.0 | 87 | 10.1 | 0.8 | 1.2 | 1.7 | ||
| Downstream | S-7 | 96.9 | 208.0 | 23.1 | 106.8 | 16.3 | 0.5 | 18.7 | 2.2 | 21.3 | 5.2 | 15.2 | 2.1 | 11.0 | 1.8 | 529 | 0.1 | 1.0 | 1.1 | 1.1 | |
| S-8 | 13.9 | 24.7 | 3.2 | 8.1 | 5.0 | 2.3 | 6.2 | 0.5 | 2.4 | 0.9 | 1.9 | 0.8 | 4.1 | 0.5 | 74 | 1.8 | 0.8 | 4.9 | 5.4 | ||
| S-9 | 10.1 | 20.1 | 2.9 | 5.1 | 2.2 | 0.8 | 1.8 | 0.5 | 3.0 | 0.9 | 1.0 | 0.4 | 2.1 | 0.5 | 51 | 1.8 | 0.8 | 2.2 | 4.4 | ||
| S-10 | 44.9 | 98.1 | 10.7 | 28.4 | 9.2 | 6.8 | 5.8 | 0.8 | 3.9 | 1.0 | 3.9 | 0.7 | 2.9 | 1.1 | 218 | 4.1 | 1.0 | 1.3 | 1.1 | ||
| Deep | Upstream | D-1 | 11.1 | 23.1 | 3.2 | 9.1 | 2.9 | 0.5 | 3.8 | 1.0 | 1.9 | 1.7 | 1.0 | 0.5 | 1.9 | 0.5 | 62 | 0.6 | 0.8 | 2.7 | 2.2 |
| D-2 | 8.2 | 20.8 | 2.7 | 7.2 | 2.9 | 0.5 | 2.3 | 0.5 | 1.7 | 0.9 | 1.2 | 0.5 | 1.5 | 0.5 | 51 | 0.9 | 1.0 | 2.0 | 2.3 | ||
| D-3 | 19.9 | 43.1 | 5.2 | 18.8 | 5.1 | 2.7 | 6.9 | 0.9 | 4.1 | 1.8 | 2.8 | 0.5 | 3.9 | 1.0 | 117 | 2.0 | 0.9 | 2.3 | 2.2 | ||
| D-4 | 15.0 | 37.1 | 4.9 | 9.2 | 3.0 | 0.5 | 4.9 | 1.1 | 3.8 | 1.8 | 4.1 | 0.4 | 1.9 | 1.1 | 89 | 0.5 | 0.9 | 3.4 | 2.2 | ||
| D-5 | 13.1 | 29.3 | 4.1 | 11.1 | 2.4 | 1.7 | 4.1 | 0.5 | 3.2 | 2.1 | 4.1 | 0.5 | 2.0 | 0.9 | 79 | 2.4 | 0.9 | 2.3 | 1.9 | ||
| Downstream | D-6 | 14.0 | 38.0 | 4.9 | 11.0 | 4.1 | 2.0 | 7.0 | 1.0 | 4.9 | 3.1 | 3.0 | 1.0 | 1.9 | 0.5 | 96 | 1.7 | 1.0 | 4.0 | 1.8 | |
| D-7 | 13.0 | 39.1 | 3.9 | 12.1 | 2.2 | 1.1 | 4.1 | 0.5 | 2.2 | 2.5 | 1.2 | 0.5 | 1.5 | 0.4 | 84 | 1.6 | 1.2 | 2.2 | 1.3 | ||
| D-8 | 18.1 | 42.8 | 4.2 | 7.9 | 3.9 | 0.5 | 4.5 | 1.0 | 5.0 | 2.8 | 3.0 | 0.5 | 2.5 | 0.5 | 97 | 0.5 | 1.1 | 3.6 | 3.4 | ||
| D-9 | 10.3 | 28.9 | 4.1 | 6.2 | 3.1 | 0.5 | 4.0 | 0.5 | 3.2 | 2.8 | 2.0 | 0.5 | 1.0 | 0.5 | 68 | 0.6 | 1.0 | 4.1 | 1.7 | ||
| D-10 | 15.2 | 35.0 | 3.2 | 11.0 | 4.9 | 0.5 | 4.2 | 0.5 | 2.1 | 1.1 | 2.0 | 0.5 | 1.5 | 0.5 | 82 | 0.5 | 1.1 | 2.4 | 1.4 | ||
| D-11 | 13.2 | 32.1 | 4.8 | 8.2 | 2.1 | 0.5 | 4.0 | 0.5 | 2.1 | 0.9 | 2.0 | 0.5 | 1.9 | 0.5 | 73 | 0.8 | 0.9 | 3.1 | 2.5 | ||
| D-12 | 23.2 | 55.2 | 7.3 | 19.0 | 3.0 | 1.1 | 6.0 | 1.1 | 3.0 | 1.0 | 3.0 | 0.5 | 2.2 | 0.5 | 126 | 1.1 | 0.9 | 2.0 | 1.2 | ||
| D-13 | 41.3 | 90.1 | 10.2 | 42.1 | 9.3 | 2.5 | 9.4 | 1.5 | 8.4 | 2.1 | 4.6 | 0.8 | 4.9 | 0.8 | 228 | 1.2 | 1.0 | 1.4 | 1.2 | ||
References
- Nelson, B.J.; Wood, S.A.; Osiensky, J.L. Rare earth element geochemistry of groundwater in the Palouse Basin, northern Idaho-eastern Washington. Geochem. Explor. Environ. Anal. 2004, 4, 227–241. [Google Scholar] [CrossRef]
- Dia, A.; Gruau, G.; Olivié-Lauquet, G.; Riou, C.; Molénat, J.; Curmi, P. The distribution of rare earth elements in groundwaters: Assessing the role of source-rock composition, redox changes and colloidal particle. Geochim. Cosmochim. Acta 2000, 64, 4131–4151. [Google Scholar] [CrossRef]
- Gruau, G.; Dia, A.; Olivié-Lauquet, G.; Davranche, M.; Pinay, G. Controls on the distribution of rare earth elements in shallow groundwaters. Water Res. 2004, 38, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, B.; Wang, G.; Shen, Z. Geochemical controls on arsenic and rare earth elements approximately along a groundwater flow path in the shallow aquifer of the Hetao Basin, Inner Mongolia. Chem. Geol. 2010, 270, 117–125. [Google Scholar] [CrossRef]
- Pourret, O.; Gruau, G.; Dia, A.; Davranche, M.; Molénat, J. Colloidal control on the distribution of rare earth elements in shallow groundwaters. Aquat. Geochem. 2010, 16, 31–59. [Google Scholar] [CrossRef]
- Smedley, P.L. The geochemistry of rare earth elements in groundwater from the Carnmenellis area, southwest England. Geochim. Cosmochim. Acta 1991, 55, 2767–2779. [Google Scholar] [CrossRef]
- Noack, C.W.; Dzombak, D.A.; Karamalidis, A.K. Rare earth element distributions and trends in natural waters with a focus on groundwater. Environ. Sci. Technol. 2014, 48, 4317–4326. [Google Scholar] [CrossRef]
- German, C.R.; Elderfield, H. Application of the Ce anomaly as a paleoredox indicator: The ground rules. Paleoceanography 1990, 5, 823–833. [Google Scholar] [CrossRef]
- Willis, S.S.; Johannesson, K.H. Controls on the geochemistry of rare earth elements in sediments and groundwaters of the Aquia aquifer, Maryland, USA. Chem. Geol. 2011, 285, 32–49. [Google Scholar] [CrossRef]
- Liu, H.; Pourret, O.; Guo, H.; Bonhoure, J. Rare earth elements sorption to iron oxyhydroxide: Model development and application to groundwater. Appl. Geochem. 2017, 87 (Suppl. C), 158–166. [Google Scholar] [CrossRef]
- Merschel, G.; Bau, M.; Dantas, E.L. Contrasting impact of organic and inorganic nanoparticles and colloids on the behavior of particle-reactive elements in tropical estuaries: An experimental study. Geochim. Cosmochim. Acta 2017, 197, 1–13. [Google Scholar] [CrossRef]
- Pourret, O.; Tuduri, J. Continental shelves as potential resource of rare earth elements. Sci. Rep. 2017, 7, 5857. [Google Scholar] [CrossRef] [PubMed]
- Verplanck, P.L.; Nordstrom, D.K.; Taylor, H.E.; Kimball, B.A. Rare earth element partitioning between hydrous ferric oxides and acid mine water during iron oxidation. Appl. Geochem. 2004, 19, 1339–1354. [Google Scholar] [CrossRef]
- Quinn, K.A.; Byrne, R.H.; Schijf, J. Sorption of yttrium and rare earth elements by amorphous ferric hydroxide: Influence of pH and ionic strength. Mar. Chem. 2006, 99, 128–150. [Google Scholar] [CrossRef]
- Leybourne, M.I.; Goodfellow, W.D.; Boyle, D.R.; Hall, G.M. Rapid development of negative Ce anomalies in surface waters and contrasting REE patterns in groundwaters associated with Zn-Pb massive sulphide deposits. Appl. Geochem. 2000, 15, 695–723. [Google Scholar] [CrossRef]
- Bau, M.; Koschinsky, A. Oxidative scavenging of cerium on hydrous Fe oxide: Evidence from the distribution of rare earth elements and yttrium between Fe oxides and Mn oxides in hydrogenetic ferromanganese crusts. Geochem. J. 2009, 43, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Josso, P.; Pelleter, E.; Pourret, O.; Fouquet, Y.; Etoubleau, J.; Cheron, S.; Bollinger, C. A new discrimination scheme for oceanic ferromanganese deposits using high field strength and rare earth elements. Ore Geol. Rev. 2017, 87, 3–15. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Wang, X.; Ren, Z.; Li, L. Salt-Water Intrusion in the Lower Reaches of the Weihe River, Shandong Province, China. Hydrogeol. J. 1997, 5, 82–88. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Shi, D.H.; Shen, Z.L.; Zhong, Z.S.; Xue, Y.Q. Evolution and development of groundwater environment in North China Plain under human activities. Acta Geosic. Sin. 1997, 18, 337–344. [Google Scholar]
- Liu, H.; Guo, H.; Xing, L.; Zhan, Y.; Li, F.; Shao, J.; Niu, H.; Liang, X.; Li, C. Geochemical behaviors of rare earth elements in groundwater along a flow path in the North China Plain. J. Asian Earth Sci. 2016, 117, 33–51. [Google Scholar] [CrossRef]
- Chen, W.H. Groundwater in Hebei; Seismological Press: Beijing, China, 1999. (In Chinese) [Google Scholar]
- Han, D.M.; Song, X.F.; Currell, M.J.; Tsujimura, M. Using chlorofluorocarbons (CFCs) and tritium to improve conceptual model of groundwater flow in the South Coast Aquifers of Laizhou Bay, China. Hydrol. Process. 2012, 26, 3614–3629. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Techniques and Methods; U.S. Geological Survey: Denver, CO, USA, 2013.
- Hummel, W.; Berner, U.; Curti, E.; Pearson, F.J.; Thoenen, T. Nagra/PSI Chemical Thermodynamic Data Base 01/01. Radiochim. Acta 2002, 90, 805–813. [Google Scholar] [CrossRef]
- Luo, Y.; Millero, F.J. Effects of temperature and ionic strength on the stabilities of the first and second fluoride complexes of yttrium and the rare earth elements. Geochim. Cosmochim. Acta 2004, 68, 4301–4308. [Google Scholar] [CrossRef]
- Luo, Y.R.; Byrne, R.H. Yttrium and rare Earth element complexation by chloride ions at 25 °C. J. Solut. Chem. 2001, 30, 837–845. [Google Scholar] [CrossRef]
- Luo, Y.R.; Byrne, R.H. Carbonate complexation of yttrium and the rare earth elements in natural waters. Geochim. Cosmochim. Acta 2004, 68, 691–699. [Google Scholar] [CrossRef]
- Klungness, G.D.; Byrne, R.H. Comparative hydrolysis behavior of the rare earths and yttrium: The influence of temperature and ionic strength. Polyhedron 2000, 19, 99–107. [Google Scholar] [CrossRef]
- Millero, F.J. Stability constants for the formation of rare earth inorganic complexes as a function of ionic strength. Geochim. Cosmochim. Acta 1992, 56, 3123–3132. [Google Scholar] [CrossRef]
- Pourret, O.; Davranche, M. Rare earth element sorption onto hydrous manganese oxide: A modeling study. J. Colloid Interface Sci. 2013, 395, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Schijf, J.; Byrne, R.H. Determination of SO4β1 for yttrium and the rare earth elements at I = 0.66 m and t = 25 °C-implications for YREE solution speciation in sulfate-rich waters. Geochim. Cosmochim. Acta 2004, 68, 2825–2837. [Google Scholar] [CrossRef]
- Lee, J.H.; Byrne, R.H. Examination of comparative rare earth element complexation behavior using linear free-energy relationships. Geochim. Cosmochim. Acta 1992, 56, 1127–1137. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Plummer, L.N.; Langmuir, D.; Busenberg, E.; May, H.M.; Jones, B.F.; Parkhurst, D.L. Revised Chemical Equilibrium Data for Major Water—Mineral Reactions and Their Limitations. In Chemical Modeling of Aqueous Systems II; American Chemical Society: Washington, DC, USA, 1990; Volume 416, pp. 398–413. [Google Scholar]
- Davis, J.A.; Meece, D.E.; Kohler, M.; Curtis, G.P. Approaches to surface complexation modeling of Uranium(VI) adsorption on aquifer sediments. Geochim. Cosmochim. Acta 2004, 68, 3621–3641. [Google Scholar] [CrossRef]
- Bau, M. Scavenging of dissolved yttrium and rare earths by precipitating iron oxyhydroxide: Experimental evidence for Ce oxidation, Y-Ho fractionation, and lanthanide tetrad effect. Geochim. Cosmochim. Acta 1999, 63, 67–77. [Google Scholar] [CrossRef]
- McLennan, S.M. Relationships between the trace element composition of sedimentary rocks and upper continental crust. Geochem. Geophys. Geosyst. 2001, 2, 109. [Google Scholar] [CrossRef]
- Nesbitt, H.W. Mobility and fractionation of rare earth elements during weathering of a granodiorite. Nature 1979, 279, 206–210. [Google Scholar] [CrossRef]
- Decrée, S.; Pourret, O.; Baele, J.-M. Rare earth element fractionation in heterogenite (CoOOH): Implication for cobalt oxidized ore in the Katanga Copperbelt (Democratic Republic of Congo). J. Geochem. Explor. 2015, 159, 290–301. [Google Scholar] [CrossRef]
- Bwire Ojiambo, S.; Berry Lyons, W.; Welch, K.A.; Poreda, R.J.; Johannesson, K.H. Strontium isotopes and rare earth elements as tracers of groundwater–lake water interactions, Lake Naivasha, Kenya. Appl. Geochem. 2003, 18, 1789–1805. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Elderfield, H. The oceanic chemistry of the rare earth elements in seawater. Philos. Trans. R. Soc. Lond. 1988, A325, 105–126. [Google Scholar] [CrossRef]
- Moffett, J.W. Microbially mediated cerium oxidation in sea water. Nature 1990, 345, 421–423. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Koeppenkastrop, D.; De Carlo, E.H. Uptake of rare earth elements from solution by metal oxides. Environ. Sci. Technol. 1993, 27, 1796–1802. [Google Scholar] [CrossRef]
- Bethke, C.M. Geochemical and Biogeochemical Reaction Modeling, 2nd ed.; Cambridge University Press: Cambridge, UK, 2007; pp. 1–543. ISBN 9780521875547. [Google Scholar]
- Schijf, J.; Marshall, K.S. YREE sorption on hydrous ferric oxide in 0.5M NaCl solutions: A model extension. Mar. Chem. 2011, 123, 32–43. [Google Scholar] [CrossRef]
- Elderfield, H.; Upstill-Goddard, R.; Sholkovitz, E.R. The rare earth elements in rivers, estuaries, and coastal seas and their significance to the composition of ocean waters. Geochim. Cosmochim. Acta 1990, 54, 971–991. [Google Scholar] [CrossRef]
- Marsac, R.; Real, F.; Banik, N.L.; Pedrot, M.; Pourret, O.; Vallet, V. Aqueous chemistry of Ce(IV): Estimations using actinide analogues. Dalton Trans. 2017, 46, 13553–13561. [Google Scholar] [CrossRef] [PubMed]
- Nakada, R.; Takahashi, Y.; Tanimizu, M. Isotopic and speciation study on cerium during its solid-water distribution with implication for Ce stable isotope as a paleo-redox proxy. Geochim. Cosmochim. Acta 2013, 103, 49–62. [Google Scholar] [CrossRef]
- Ohta, A.; Kawabe, I. REE(III) adsorption onto Mn dioxide and Fe oxyhydroxide: Ce(III) oxidation by Mn dioxide. Geochim. Cosmochim. Acta 2001, 65, 695–703. [Google Scholar] [CrossRef]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Pourret, O.; Guo, H.; Martinez, R.E.; Zouhri, L. Impact of Hydrous Manganese and Ferric Oxides on the Behavior of Aqueous Rare Earth Elements (REE): Evidence from a Modeling Approach and Implication for the Sink of REE. Int. J. Environ. Res. Public Health 2018, 15, 2837. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph15122837
Liu H, Pourret O, Guo H, Martinez RE, Zouhri L. Impact of Hydrous Manganese and Ferric Oxides on the Behavior of Aqueous Rare Earth Elements (REE): Evidence from a Modeling Approach and Implication for the Sink of REE. International Journal of Environmental Research and Public Health. 2018; 15(12):2837. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph15122837
Chicago/Turabian StyleLiu, Haiyan, Olivier Pourret, Huaming Guo, Raul E. Martinez, and Lahcen Zouhri. 2018. "Impact of Hydrous Manganese and Ferric Oxides on the Behavior of Aqueous Rare Earth Elements (REE): Evidence from a Modeling Approach and Implication for the Sink of REE" International Journal of Environmental Research and Public Health 15, no. 12: 2837. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph15122837