
Transcriptional Analysis of Microcystis aeruginosa Co-Cultured with Algicidal Bacteria Brevibacillus laterosporus

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cyanobacteria and Algicidal Bacterium
2.2. Measurement of the Algicidal Efficiency
2.3. Transcriptomic Samples Preparation
2.4. Transcriptomic Analysis
2.5. Quantitative Real-Time PCR Validation
3. Results
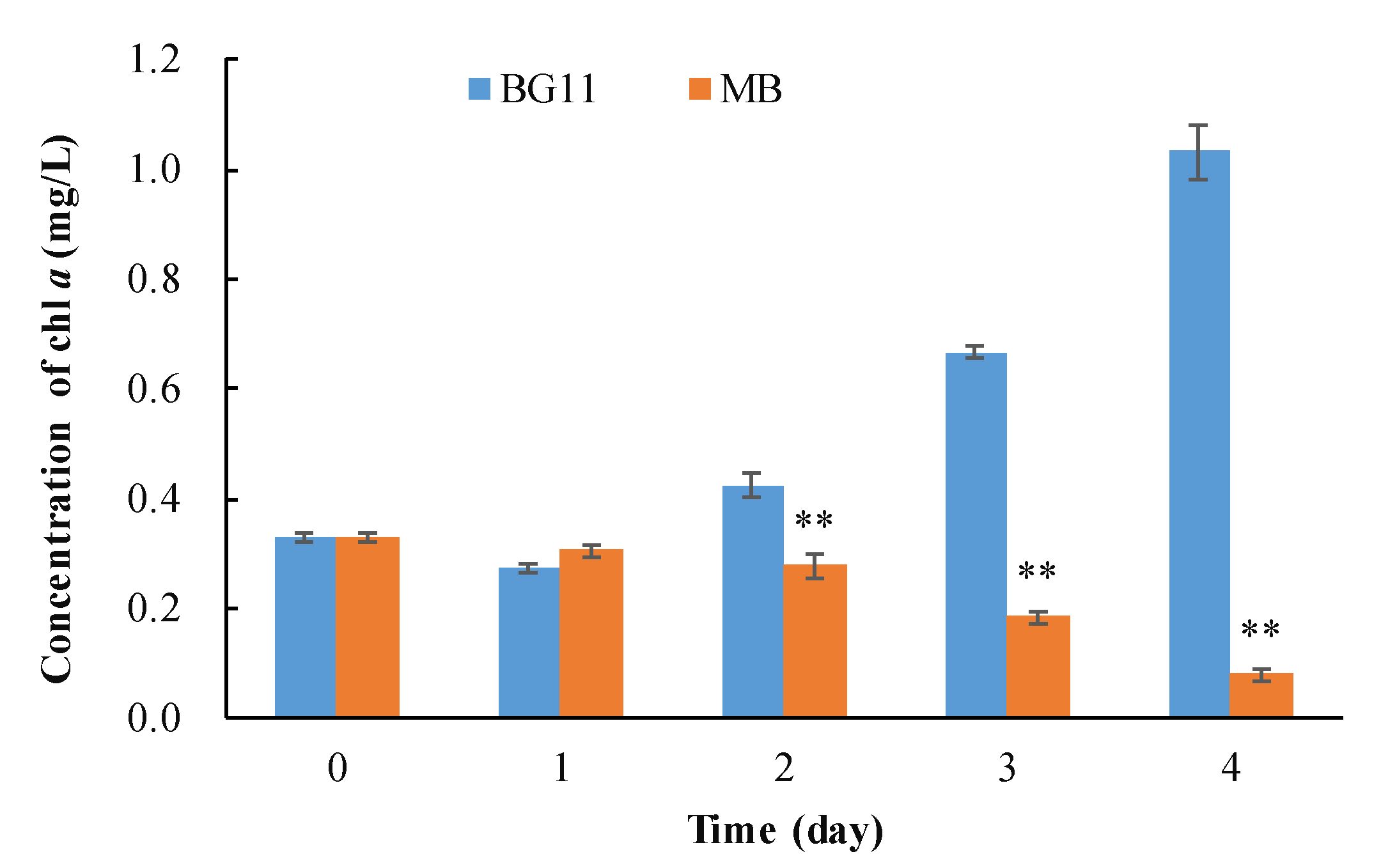
3.1. The Algicidal Efficiency
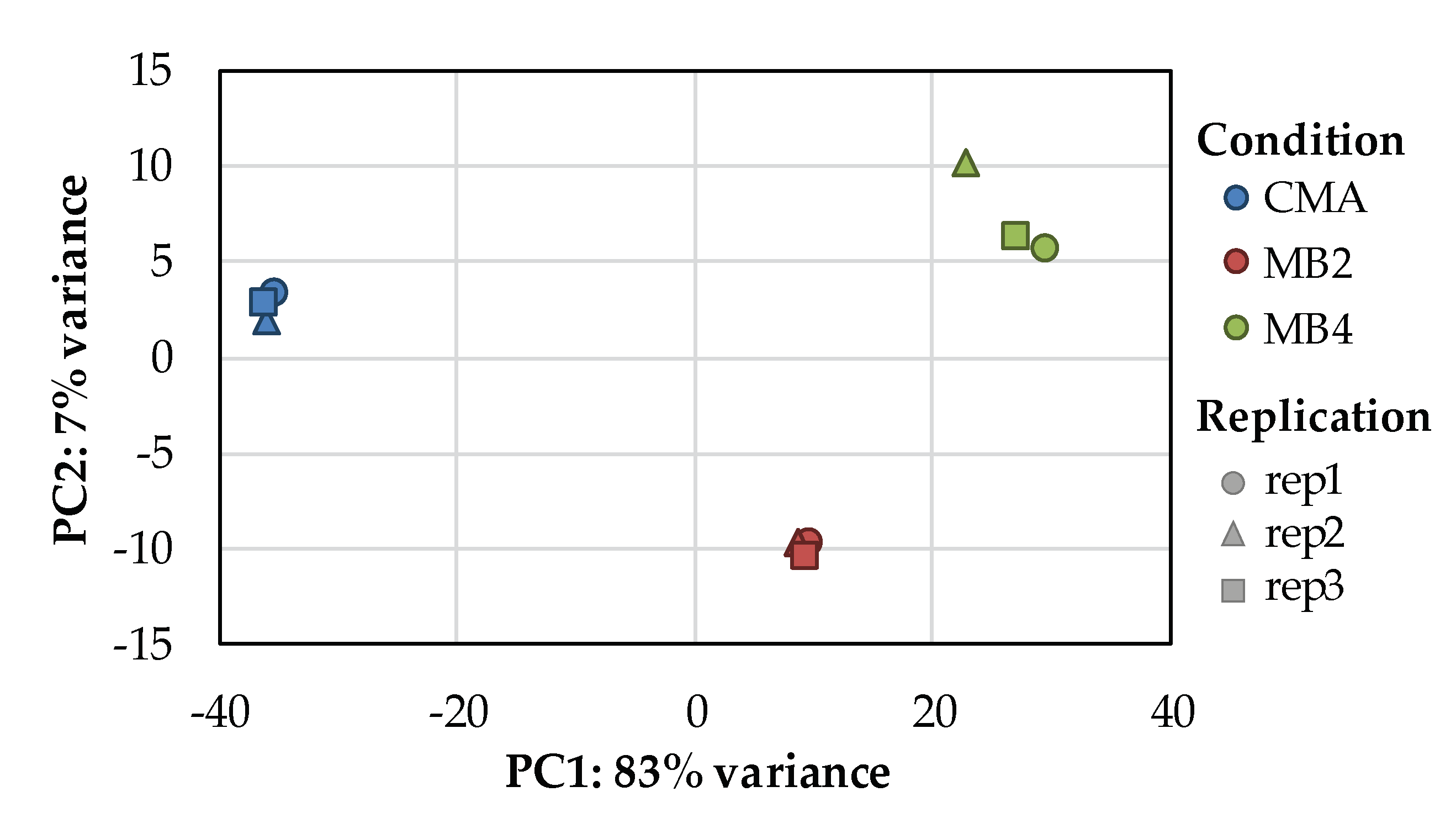
3.2. Illumina Sequencing Assembly Data Quality Analysis
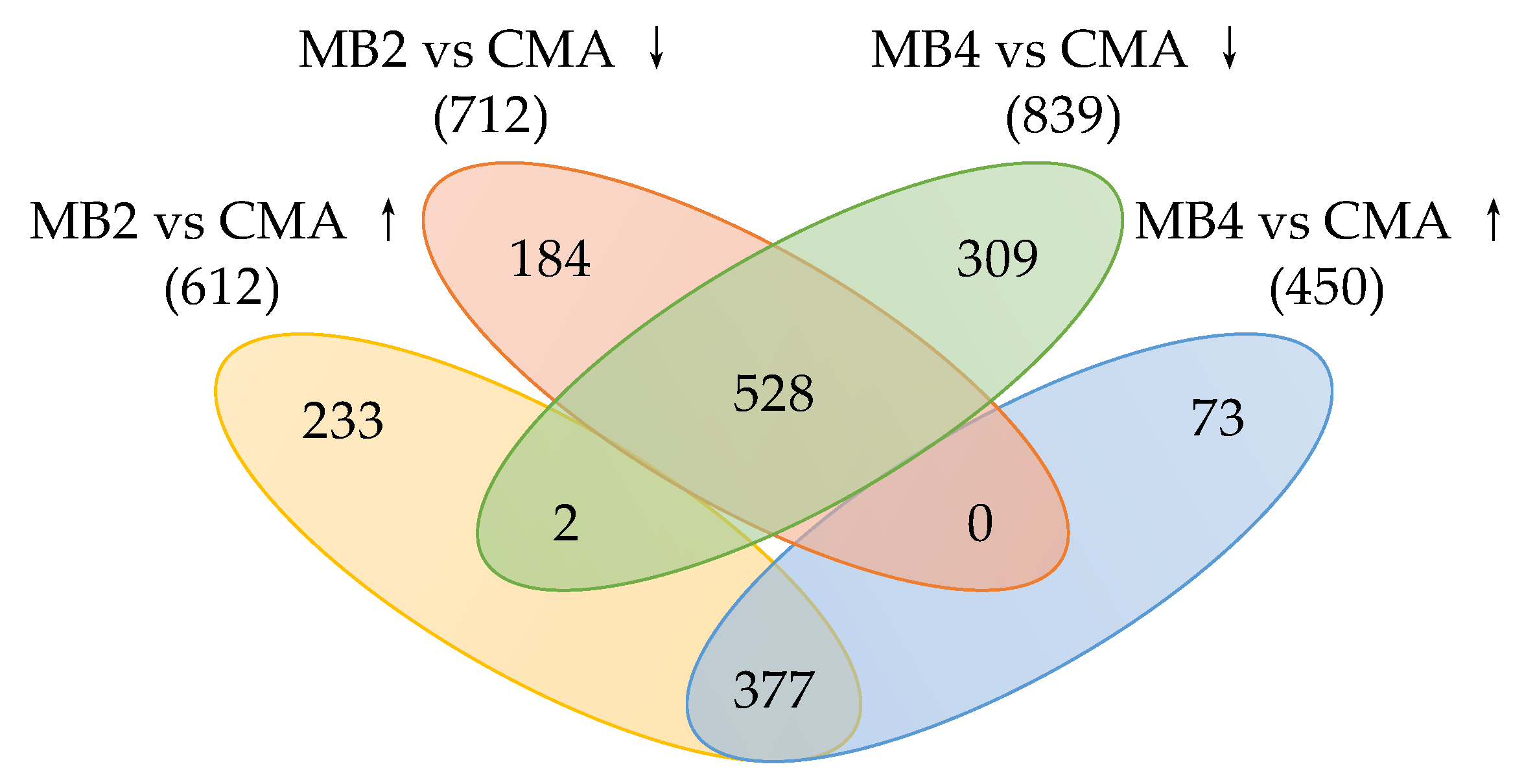
3.3. Identification of DEGs
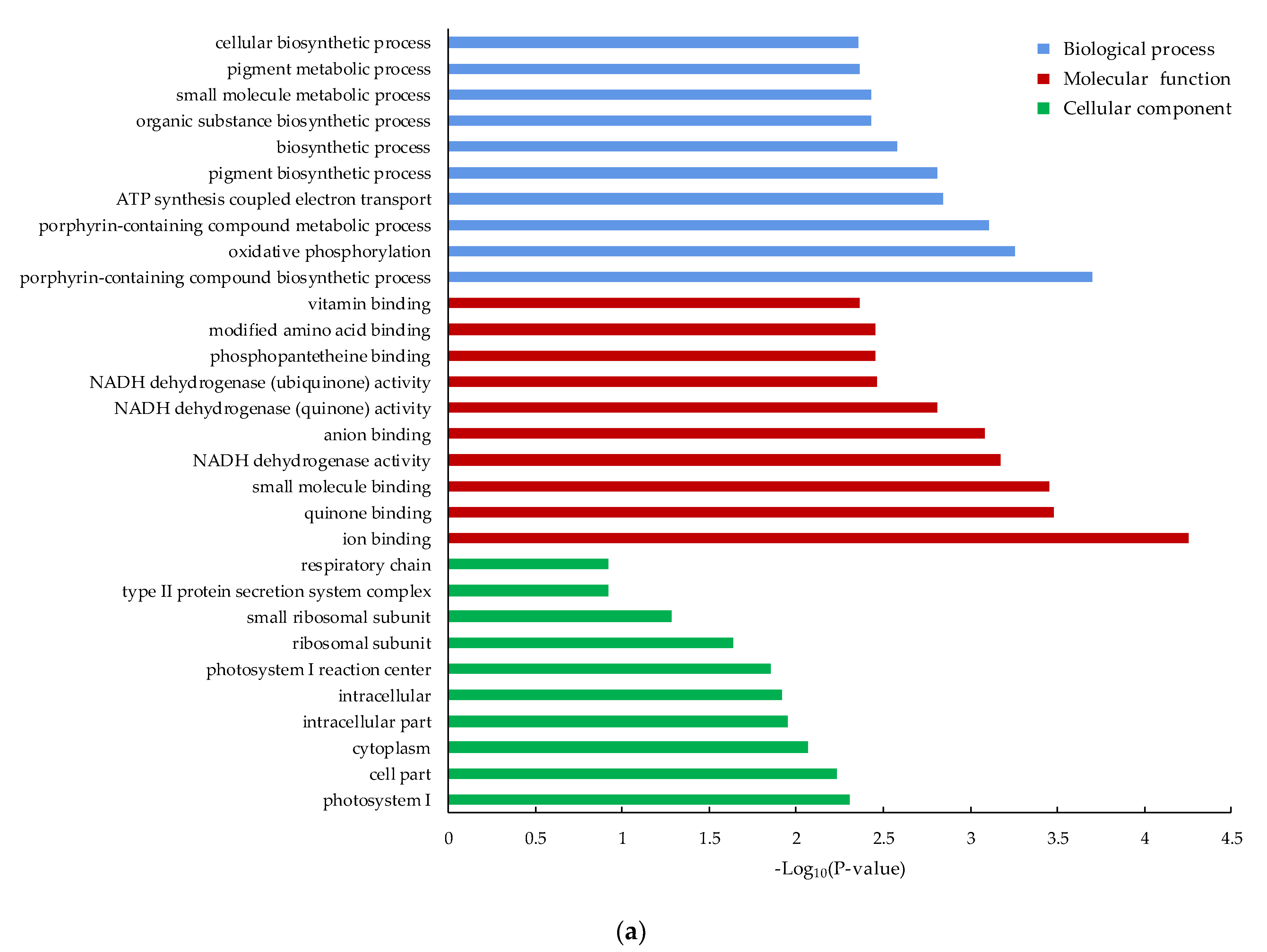
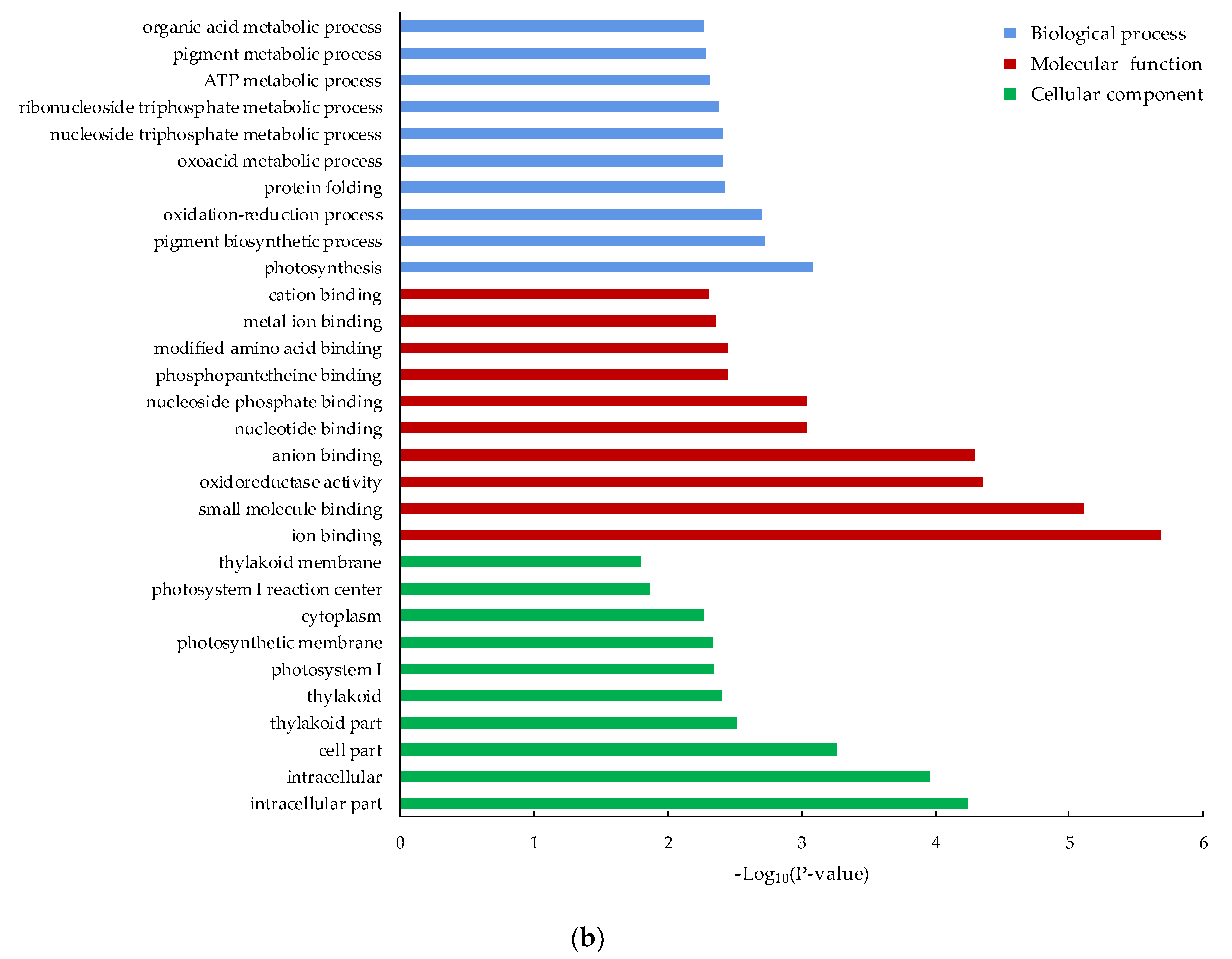
3.4. Functional Classification of the DEGs by GO and KEGG Pathway Analysis
3.5. Algicidal-Related Gene and Pathway Analysis
3.6. Quantitative Real-Time PCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Watanabe, M.F.; Oishi, S.; Watanabe, Y.; Watanabe, M. Strong Probability Of Lethal Toxicity In The Blue-Green Alga Microcystis Viridis Lemmermann1. J. Phycol. 1986, 22, 552–556. [Google Scholar] [CrossRef]
- Sivonen, K.; Carmichael, W.W.; Namikoshi, M.; Rinehart, K.L.; Dahlem, A.M.; Niemelä, S.I. Isolation and characterization of hepatotoxic microcystin homologs from the filamentous freshwater cyanobacterium Nostoc sp. strain 152. Appl. Environ. Microbiol. 1990, 56, 2650–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laub, J.; Henriksen, P.; Brittain, S.M.; Wang, J.; Carmichael, W.W.; Rinehart, K.L. [ADMAdda5]-microcystins inPlanktothrix agardhii strain PH-123 (cyanobacteria)? Importance for monitoring of microcystins in the environment. Environ. Toxicol. 2002, 17, 351–357. [Google Scholar] [CrossRef]
- Qin, B.; Zhu, G.; Gao, G.; Zhang, Y.; Li, W.; Paerl, H.W.; Carmichael, W.W. A Drinking Water Crisis in Lake Taihu, China: Linkage to Climatic Variability and Lake Management. Environ. Manag. 2010, 45, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Otten, T.G. Blooms Bite the Hand That Feeds Them. Science 2013, 342, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Harke, M.J.; Steffen, M.M.; Gobler, C.J.; Otten, T.G.; Wilhelm, S.; Wood, S.A.; Paerl, H.W. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae 2016, 54, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wu, M.; Yang, W.; Yin, W.; Jin, F.; Ye, M.; Currie, N.; Scholz, M. Ecological Strategy for Eutrophication Control. Water. Air Soil Pollut. 2011, 223, 723–737. [Google Scholar] [CrossRef]
- Sun, P.; Esquivel-Elizondo, S.; Zhao, Y.; Wu, Y. Glucose triggers the cytotoxicity of Citrobacter sp. R1 against Microcystis aeruginosa. Sci. Total. Environ. 2017, 603–604, 18–25. [Google Scholar] [CrossRef]
- Liu, J.; Yang, C.; Chi, Y.; Wu, D.; Dai, X.; Zhang, X.; Igarashi, Y.; Luo, F. Algicidal characterization and mechanism of Bacillus licheniformis Sp34 against Microcystis aeruginosa in Dianchi Lake. J. Basic Microbiol. 2019, 59, 1112–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Massey, I.Y.; Liu, Y.; Huang, F.; Gao, R.; Ding, M.; Xiang, L.; He, C.; Wei, J.; Li, Y.; et al. Identification and characterization of a novel indigenous algicidal bacterium Chryseobacterium species against Microcystis aeruginosa. J. Toxicol. Environ. Health Part A 2019, 82, 845–853. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Y.; Zhang, D.; Chen, S.; Bai, X.; Ma, X.; Xie, Z.; Xu, H. Effect and mechanism of the algicidal bacterium Sulfitobacter porphyrae ZFX1 on the mitigation of harmful algal blooms caused by Prorocentrum donghaiense. Environ. Pollut. 2020, 263, 114475. [Google Scholar] [CrossRef] [PubMed]
- Danyang, Z.; Chen, Q.; Zhang, D.; Zhang, H.; Lei, X.; Chen, Z.; Qiuliang, C.; Hong, Y.; Ma, X.; Zheng, W.; et al. The algicidal mechanism of prodigiosin from Hahella sp. KA22 against Microcystis aeruginosa. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhou, Y.; Yin, L.; Zhu, G.; Liang, G.; Pu, Y. Microcystin-Degrading Activity of an Indigenous Bacterial Strain Stenotrophomonas acidaminiphila MC-LTH2 Isolated from Lake Taihu. PLoS ONE 2014, 9, e86216. [Google Scholar] [CrossRef] [Green Version]
- Shilo, M. Lysis of Blue-Green Algae by Myxobacter. J. Bacteriol. 1970, 104, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, G.; Yoshikawa, T.; Yasuda, A.; Sakata, T. Algicidal activity and gliding motility of Saprospira sp. SS98-5. Can. J. Microbiol. 2003, 49, 92–100. [Google Scholar] [CrossRef]
- Hou, S.; Shu, W.; Tan, S.; Zhao, L.; Yin, P. Exploration of the antioxidant system and photosynthetic system of a marine algicidal Bacillus and its effect on four harmful algal bloom species. Can. J. Microbiol. 2016, 62, 49–59. [Google Scholar] [CrossRef]
- Zhang, X.; Song, T.; Ma, H.; Li, L. Correction: Physiological response of Microcystis aeruginosa to the extracellular substances from an Aeromonas sp. RSC Adv. 2017, 7, 51373. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Tang, D.; Liu, Y. Effects of an Algicidal Bacterium Pseudomonas mendocina on the Growth and Antioxidant System of Aphanizomenon flos-aquae. Curr. Microbiol. 2009, 59, 107–112. [Google Scholar] [CrossRef]
- Wang, H.; Tomasch, J.; Jarek, M.; Wagner-Döbler, I. A dual-species co-cultivation system to study the interactions between Roseobacters and dinoflagellates. Front. Microbiol. 2014, 5, 311. [Google Scholar] [CrossRef]
- Gao, X.; Wang, C.; Dai, W.; Ren, S.; Tao, F.; He, X.; Han, G.; Wang, W. Proteomic analysis reveals large amounts of decomposition enzymes and major metabolic pathways involved in algicidal process of Trametes versicolor F21a. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.J.; Rabinovitch, L.; Monnerat, R.G.; Passos, L.K.J.; Zahner, V. Molecular Characterization of Brevibacillus laterosporus and Its Potential Use in Biological Control. Appl. Environ. Microbiol. 2004, 70, 6657–6664. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Huang, X.; Li, C.; Zhang, N.; Wei, H.; Zhang, Y. Effects of Brevibacillus laterosporus on microbial community of Microcystis aeruginosa. J. Guangdong Ocean Univ. 2021, 41, 1–9. [Google Scholar]
- Zhang, Y.; Chen, D.; Cai, J.; Zhang, N.; Li, F.; Li, C.; Huang, X. Complete Genome Sequence Analysis of Brevibacillus laterosporus Bl-zj Reflects its Potential Algicidal Response. Curr. Microbiol. 2021, 78, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Rippka, R.; Stanier, R.Y.; Deruelles, J.; Herdman, M.; Waterbury, J.B. Generic Assignments, Strain Histories and Properties of Pure Cultures of Cyanobacteria. Microbiology 1979, 111, 1–61. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, S.; Humphrey, G. New spectrophotometric equations for determining chlorophylls a, b, c1 and c2 in higher plants, algae and natural phytoplankton. Biochem. Physiol. Pflanz. 1975, 167, 191–194. [Google Scholar] [CrossRef]
- Wang, T.; Hu, Y.; Zhu, M.; Yin, S. Integrated transcriptome and physiology analysis of Microcystis aeruginosa after exposure to copper sulfate. J. Oceanol. Limnol. 2019, 38, 102–113. [Google Scholar] [CrossRef]
- Croce, R.; Amerongen, H.V. Light-harvesting in photosystem I. Photosynth. Res. 2013, 116, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Amunts, A.; Drory, O.; Nelson, N. The structure of a plant photosystem I supercomplex at 3.4 Å resolution. Nat. Cell Biol. 2007, 447, 58–63. [Google Scholar] [CrossRef]
- Germano, M.; Yakushevska, A.E.; Keegstra, W.; Van Gorkom, H.J.; Dekker, J.P.; Boekema, E.J. Supramolecular organization of photosystem I and light-harvesting complex I in Chlamydomonas reinhardtii. FEBS Lett. 2002, 525, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Lushy, A.; Verchovsky, L.; Nechushtai, R. The Stable Assembly of Newly Synthesized PsaE into the Photosystem I Complex Occurring via the Exchange Mechanism Is Facilitated by Electrostatic Interactions. Biochemistry 2002, 41, 11192–11199. [Google Scholar] [CrossRef] [PubMed]
- Minai, L.; Fish, A.; Darash-Yahana, M.; Verchovsky, L.; Nechushtai, R. The Assembly of the PsaD Subunit into the Membranal Photosystem I Complex Occurs via an Exchange Mechanism. Biochemistry 2001, 40, 12754–12760. [Google Scholar] [CrossRef]
- Hippler, M.; Drepper, F.; Farah, J.; Rochaix, J.-D. Fast electron transfer from cytochrome c6 and plastocyanin to photosystem I of Chlamydomonas reinhardtii requires PsaF. Biochemistry 1997, 36, 6343–6349. [Google Scholar] [CrossRef]
- Lunde, C.; Jensen, P.E.; Haldrup, A.; Knoetzel, J.; Scheller, H. The PSI-H subunit of photosystem I is essential for state transitions in plant photosynthesis. Nat. Cell Biol. 2000, 408, 613–615. [Google Scholar] [CrossRef]
- Sakata, S.; Mizusawa, N.; Kubota-Kawai, H.; Sakurai, I.; Wada, H. Psb28 is involved in recovery of photosystem II at high temperature in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Cramer, W.A.; Soriano, G.M.; Ponomarev, M.; Huang, D.; Zhang, H.; Martinez, S.E.; Smith, J.L. Some new structural aspects and old controversies concerning the cytochrome b6f complex of oxygenic photosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 477–508. [Google Scholar] [CrossRef] [PubMed]
- Boekema, E.J.; Braun, H.-P. Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System. J. Biol. Chem. 2007, 282, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, T. Complex I: A chimaera of a redox and conformation-driven proton pump? J. Bioenerg. Biomembr. 2001, 33, 169–177. [Google Scholar] [CrossRef]
- Cardol, P.; Matagne, R.F.; Remacle, C. Impact of Mutations Affecting ND Mitochondria-encoded Subunits on the Activity and Assembly of Complex I in Chlamydomonas. Implication for the Structural Organization of the Enzyme. J. Mol. Biol. 2002, 319, 1211–1221. [Google Scholar] [CrossRef]
- Cardol, P.; Boutaffala, L.; Memmi, S.; Devreese, B.; Matagne, R.F.; Remacle, C. In Chlamydomonas, the loss of ND5 subunit prevents the assembly of whole mitochondrial complex I and leads to the formation of a low abundant 700 kDa subcomplex. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Park, J.-S.; Deng, J.-H.; Bai, Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Pierrel, F.; Bestwick, M.L.; Cobine, P.; Khalimonchuk, O.; Cricco, J.; Winge, D.R. Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 2007, 26, 4335–4346. [Google Scholar] [CrossRef]
- Yoshida, M.; Muneyuki, E.; Hisabori, T. ATP synthase—A marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Chen, Q. Transcriptional Analysis of Chlorella Pyrenoidosa Exposed to Bisphenol A. Int. J. Environ. Res. Public Health 2019, 16, 1374. [Google Scholar] [CrossRef] [Green Version]
- Guschina, I.A.; Harwood, J.L. Lipids and lipid metabolism in eukaryotic algae. Prog. Lipid Res. 2006, 45, 160–186. [Google Scholar] [CrossRef] [PubMed]
- Ruangsomboon, S.; Ganmanee, M.; Choochote, S. Effects of different nitrogen, phosphorus, and iron concentrations and salinity on lipid production in newly isolated strain of the tropical green microalga, Scenedesmus dimorphus KMITL. Environ. Boil. Fishes 2012, 25, 867–874. [Google Scholar] [CrossRef]
- Pandit, P.R.; Fulekar, M.H.; Karuna, M.S.L. Effect of salinity stress on growth, lipid productivity, fatty acid composition, and biodiesel properties in Acutodesmus obliquus and Chlorella vulgaris. Environ. Sci. Pollut. Res. 2017, 24, 13437–13451. [Google Scholar] [CrossRef]
- Allen, E.E.; Bartlett, D.H. FabF Is Required for Piezoregulation of cis -Vaccenic Acid Levels and Piezophilic Growth of the Deep-Sea Bacterium Photobacterium profundum Strain SS9. J. Bacteriol. 2000, 182, 1264–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Gonsaud, M.; Ducasse, S.; Hoh, F.; Zerbib, D.; Labesse, G.; Quemard, A. Crystal Structure of MabA from Mycobacterium tuberculosis, a Reductase involved in Long-chain Fatty Acid Biosynthesis. J. Mol. Biol. 2002, 320, 249–261. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Cronan, J.E. β-Ketoacyl-Acyl Carrier Protein Synthase III (FabH) Is Essential for Bacterial Fatty Acid Synthesis. J. Biol. Chem. 2003, 278, 51494–51503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, R.J.; Rock, C.O. Roles of the FabA and FabZ β-Hydroxyacyl-Acyl Carrier Protein Dehydratases in Escherichia coli Fatty Acid Biosynthesis. J. Biol. Chem. 1996, 271, 27795–27801. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Q.; Feng, J.; Lv, J.-P.; Xie, S.-L. Effect of high-doses pyrogallol on oxidative damage, transcriptional responses and microcystins synthesis in Microcystis aeruginosa TY001 (Cyanobacteria). Ecotoxicol. Environ. Saf. 2016, 134, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Stork, T.; Michel, K.-P.; Pistorius, E.K.; Dietz, K.-J. Bioinformatic analysis of the genomes of the cyanobacteria Synechocystis sp. PCC 6803 and Synechococcus elongatus PCC 7942 for the presence of peroxiredoxins and their transcript regulation under stress. J. Exp. Bot. 2005, 56, 3193–3206. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
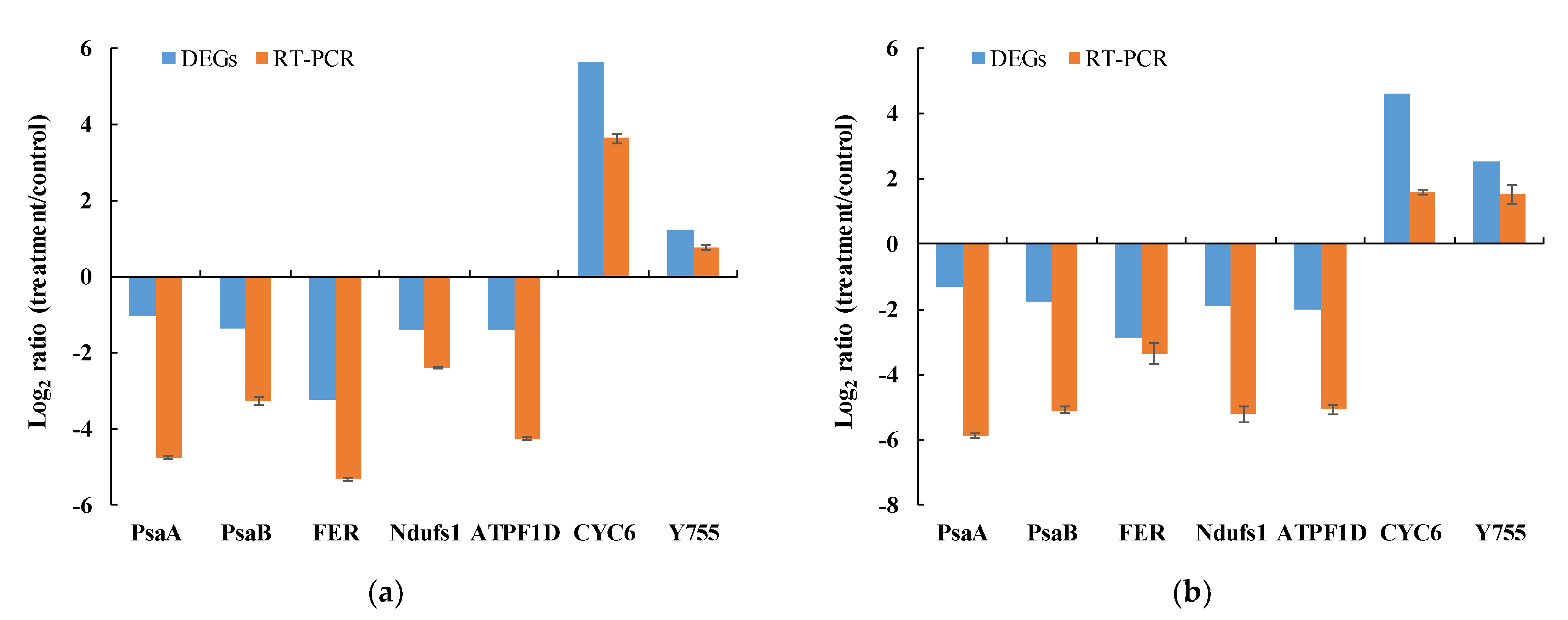{kind=link}
| Sample | Raw Data (bp) | Raw Reads No. | Clean Data (bp) | Clean Reads No. | Mapped Reads No. | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|
| CMA1 | 9,468,720,000 | 63,124,800 | 8,089,035,900 | 53,926,906 | 52,199,272 | 97.86 | 94.09 |
| CMA2 | 9,206,526,300 | 61,376,842 | 7,486,911,900 | 49,912,746 | 48,062,591 | 97.93 | 94.40 |
| CMA3 | 8,543,553,900 | 56,957,026 | 7,371,049,200 | 49,140,328 | 47,594,707 | 98.05 | 94.50 |
| MB2_1 | 11,260,710,900 | 75,071,406 | 9,110,216,700 | 60,734,778 | 43,388,415 | 97.73 | 94.22 |
| MB2_2 | 9,961,599,300 | 66,410,662 | 7,954,650,900 | 53,031,006 | 39,017,497 | 97.62 | 93.88 |
| MB2_3 | 9,498,113,100 | 63,320,754 | 7,660,962,000 | 51,073,080 | 36,364,637 | 97.65 | 93.90 |
| MB4_1 | 10,501,108,500 | 70,007,390 | 8,108,475,900 | 54,056,506 | 37,308,387 | 97.72 | 94.15 |
| MB4_2 | 9,475,501,500 | 63,170,010 | 7,402,896,600 | 49,352,644 | 4,777,919 | 97.29 | 93.41 |
| MB4_3 | 9,642,770,100 | 64,285,134 | 7,021,052,700 | 46,807,018 | 31,358,598 | 96.94 | 92.90 |
| Average | 9,728,733,733 | 64,858,225 | 7,800,583,533 | 52,003,890 | 37,785,780 | 97.64 | 93.94 |
| KEGG Pathways (Level 2) | MB2 vs. CMA | MB4 vs. CMA | ||
|---|---|---|---|---|
| Up | Down | Up | Down | |
| Amino acid metabolism | 42 | 25 | 39 | 38 |
| Carbohydrate metabolism | 59 | 40 | 52 | 60 |
| Energy metabolism | 31 | 44 | 27 | 52 |
| Metabolism of cofactors and vitamins | 33 | 15 | 28 | 23 |
| Lipid metabolism | 15 | 6 | 16 | 11 |
| Translation | 16 | 3 | 14 | 2 |
| Metabolism of other amino acids | 9 | 4 | 9 | 4 |
| Biosynthesis of other secondary metabolites | 7 | 4 | 9 | 5 |
| Nucleotide metabolism | 12 | 11 | 7 | 15 |
| Metabolism of terpenoids and polyketides | 8 | 7 | 7 | 4 |
| Replication and repair | 6 | 10 | 5 | 14 |
| Membrane transport | 4 | 8 | 2 | 9 |
| Xenobiotics biodegradation and metabolism | 5 | 2 | 3 | 3 |
| Transcription | 2 | 1 | 2 | 1 |
| Signal transduction | 1 | 4 | 1 | 3 |
| Immune system | 0 | 2 | 0 | 2 |
| Glycan biosynthesis and metabolism | 2 | 2 | 1 | 4 |
| Folding, sorting and degradation | 8 | 3 | 5 | 8 |
| Cell growth and death | 1 | 1 | 1 | 1 |
| Cellular community—prokaryotes | 1 | 7 | 1 | 8 |
| Environmental adaptation | - | - | 0 | 1 |
| Gene ID | Symbol | MB2 vs. CMA | MB4 vs. CMA | Annotation | ||
|---|---|---|---|---|---|---|
| log2FC | p-Value | log2FC | p-Value | |||
| Photosynthesis | ||||||
| RS02885 | PsaA | −1.02 | 1.66 × 10−3 | −1.29 | 1.35 × 10−3 | photosystem I core protein psaA |
| RS02890 | PsaB | −1.34 | 2.02 × 10−5 | −1.73 | 8.05 × 10−6 | photosystem I core protein psaB |
| RS22460 | PsaC | −2.5 | 1.13 × 10−15 | −3.97 | 3.25 × 10−19 | photosystem I subunit VII |
| RS24715 | PsaD | −2.93 | 2.42 × 10−23 | −4.04 | 6.13 × 10−23 | photosystem I reaction center subunit II |
| RS04855 | PsaE | −1.66 | 6.27 × 10−13 | −1.97 | 6.76 × 10−7 | photosystem I reaction center subunit IV |
| RS02775 | PsaF | −1.61 | 3.01 × 10−13 | −1.96 | 8.76 × 10−8 | photosystem I subunit III |
| RS25020 | PsaL | −3.61 | 1.36 × 10−53 | −3.71 | 3.78 × 10−19 | photosystem I reaction center subunit XI |
| RS13445 | PC | −4.00 | 8.78 × 10−65 | −3.40 | 3.01 × 10−22 | plastocyanin |
| RS12310 | FER | −3.24 | 4.87 × 10−29 | −2.88 | 8.76 × 10−13 | ferredoxin |
| RS15355 | Psb28 | −1.12 | 2.85 × 10−2 | - | - | photosystem II reaction center protein Psb28 |
| RS02780 | PsaJ | −1.61 | 4.15 × 10−6 | - | - | photosystem I reaction center subunit IX |
| RS15435 | UCRI | - | - | −1.6 | 9.44 × 10−5 | cytochrome b6-f complex iron-sulfur subunit 1 |
| RS16845 | ISIA | 3.94 | 1.97 × 10−61 | 3.36 | 6.36 × 10−21 | iron stress-induced chlorophyll-binding protein |
| RS13450 | CYC6 | 5.63 | 2.69 × 10−17 | 4.63 | 2.31 × 10−4 | cytochrome c6 |
| RS12320 | PsbU | - | - | 1.17 | 2.53 × 10−3 | photosystem II 12 kDa extrinsic protein |
| RS16660 | PsbJ | 1.08 | 1.00 × 10−4 | - | - | photosystem II reaction center protein J |
| Oxidative phosphorylation | ||||||
| RS07425 | ND1 | −1.38 | 7.03 × 10−9 | −1.77 | 2.21 × 10−4 | NAD(P)H-quinone oxidoreductase subunit 1 |
| RS05160 | ND2 | −1.09 | 3.98 × 10−7 | −1.72 | 5.03 × 10−6 | NAD(P)H-quinone oxidoreductase subunit 2 |
| RS13045 | ND4 | −2.57 | 3.58 × 10−10 | −2.61 | 1.02 × 10−5 | proton-translocating NADH-quinone oxidoreductase, chain M family protein |
| RS13470 | ND5 | −2.38 | 1.77 × 10−16 | −3.38 | 1.70 × 10−7 | NAD(P)H dehydrogenase, subunit NdhF3 family protein |
| RS08000 | Ndufs1 | −1.39 | 4.18 × 10−6 | −1.89 | 2.86 × 10−6 | 2Fe-2S iron-sulfur cluster binding domain protein |
| RS07420 | Ndufs8 | −2.64 | 3.79 × 10−14 | −3.43 | 6.95 × 10−15 | NADH-plastoquinone oxidoreductase, I subunit |
| RS23775 | COX10 | −1.35 | 6.50 × 10−9 | −2.27 | 2.37 × 10−11 | protoheme IX farnesyltransferase |
| RS09740 | ATPF1D | −1.39 | 1.65 × 10−4 | −2.01 | 1.96 × 10−3 | ATP synthase F1, delta subunit |
| RS09735 | ATPF0B | −1.57 | 1.47 × 10−6 | −2.33 | 1.05 × 10−7 | ATP synthase F0, B subunit |
| RS24375 | ND3 | −2.33 | 1.35 × 10−3 | - | - | NADH dehydrogenase subunit A |
| RS05535 | Ndufv2 | −1.12 | 1.16 × 10−6 | - | - | respiratory-chain NADH dehydrogenase 24 kDa subunit |
| RS23755 | COX3 | - | - | −1.64 | 9.83 × 10−4 | cytochrome c oxidase subunit III |
| RS09750 | ATPG | - | - | −1.5 | 1.37 × 10−3 | ATP synthase gamma chain |
| RS12325 | NADB | 1.5 | 3.14 × 10−7 | - | - | L-aspartate oxidase |
| RS23760 | COX1 | 1.37 | 7.16 × 10−9 | - | - | cytochrome c oxidase, subunit I |
| RS23765 | COX2 | 1.1 | 2.64 × 10−7 | - | - | cytochrome c oxidase, subunit II |
| RS24380 | Ndufs7 | - | - | 1.45 | 9.08 × 10−5 | NAD(P)H-quinone oxidoreductase subunit K 1 |
| RS13440 | DHSA | - | - | 1.53 | 9.29 × 10−5 | succinate dehydrogenase/fumarate reductase, flavoprotein subunit |
| Fatty acid biosynthesis | ||||||
| RS04585 | FabH | 1.88 | 3.38 × 10−11 | 1.97 | 1.56 × 10−2 | 3-oxoacyl-[acyl-carrier-protein] synthase III |
| RS18070 | FabG2 | 1.31 | 2.24 × 10−9 | 1.33 | 1.12 × 10−2 | PHA-specific acetoacetyl-CoA reductase |
| RS08035 | FabZ | 2.42 | 4.01 × 10−13 | 2.51 | 6.76 × 10−5 | beta-hydroxyacyl-(acyl-carrier-protein) dehydratase |
| RS22350 | FabF | 1.32 | 6.66 × 10−4 | - | - | beta-ketoacyl synthase, C-terminal domain protein |
| RS12750 | FabG1 | 1.16 | 9.88 × 10−5 | - | - | 3-oxoacyl-[acyl-carrier-protein] reductase |
| RS05845 | YOXD | - | - | 2.26 | 3.16 × 10−9 | short chain dehydrogenase family protein |
| RS23000RS09855 | AAE16 | −2.73 | 8.26 × 10−10 | −4.54 | 3.16 × 10−11 | AMP-binding enzyme family protein |
| FabI | - | - | −1.79 | 4.63 × 10−3 | enoyl-[acyl-carrier-protein] reductase [NADH] | |
| Antioxidase | ||||||
| RS02415 | Y755 | 1.21 | 2.98 × 10−8 | 2.52 | 4.86 × 10−12 | 2-Cys peroxiredoxin BAS1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Chen, D.; Zhang, N.; Li, F.; Luo, X.; Li, Q.; Li, C.; Huang, X. Transcriptional Analysis of Microcystis aeruginosa Co-Cultured with Algicidal Bacteria Brevibacillus laterosporus. Int. J. Environ. Res. Public Health 2021, 18, 8615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168615
Zhang Y, Chen D, Zhang N, Li F, Luo X, Li Q, Li C, Huang X. Transcriptional Analysis of Microcystis aeruginosa Co-Cultured with Algicidal Bacteria Brevibacillus laterosporus. International Journal of Environmental Research and Public Health. 2021; 18(16):8615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168615
Chicago/Turabian StyleZhang, Yulei, Dong Chen, Ning Zhang, Feng Li, Xiaoxia Luo, Qianru Li, Changling Li, and Xianghu Huang. 2021. "Transcriptional Analysis of Microcystis aeruginosa Co-Cultured with Algicidal Bacteria Brevibacillus laterosporus" International Journal of Environmental Research and Public Health 18, no. 16: 8615. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168615