
Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes


1
Physics Department, College of Science, Jouf University, Sakakah 11942, Saudi Arabia
2
Department of Physics, College of Science and Humanities in Al-Kharj, Prince Sattam bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
3
Department of Physics, Lancaster University, Lancaster LA1 4YB, UK
*
Author to whom correspondence should be addressed.
Energies 2023, 16(11), 4342; https://0-doi-org.brum.beds.ac.uk/10.3390/en16114342
Submission received: 9 February 2023
/
Revised: 13 April 2023
/
Accepted: 28 April 2023
/
Published: 26 May 2023
(This article belongs to the Special Issue Advanced Studies of Thermoelectric Systems)
Abstract
:A considerable potential advantage of manufacturing electric and thermoelectric devices using endohedral metallofullerenes (EMFs) is their ability to accommodate metallic moieties inside their cavities. Published experimental and theoretical works have explained the usefulness of this resilience feature for improving the electrical conductance and thermopower. Through thorough theoretical investigations of three EMF complexes employing three different metallic moieties involving Sc3C2, Sc3N, and Er3N and their configurations on a gold (111) surface, this research demonstrates that the thermoelectric properties of these molecular complexes can be tuned by taking advantage of the charge transfer from metallic moieties to Ih-C80 cages. Mulliken, Hirshfeld, and Voronoi simulations articulate that the charge migrates from metallic moieties to cages; however, the amount of the transferred charge depends on the nature of the moiety within the complex.
1. Introduction
Charge transfer (CT), electron transfer (ET), and donor–acceptor (DA) complexes have long been the focus of investigation. Consequently, charge transfer perception is essential in many organic devices, because of its various uses in many disciplines involving chemistry, physics, materials science, medicine, and biology. For example, CT has been extensively explored in organic solar cells [1,2,3], water splitting devices, [4], and single molecule electronics [5,6,7,8,9]. Similarly, there have been varying types of donors and acceptors in complicated charge transfer research [10,11,12,13]. The chemical nature of the molecule determines whether a molecule behaves as a donor or acceptor. In such systems, an electron-rich donor commonly acts as the receptor and the acceptor is often electron-deficient. In measurement methods, the charge transfer through molecular systems is classified into two categories: CT in Donor–Bridge–Acceptor (DBA) molecules and CT in Metal–Bridge–Metal (MBM) junctions [14,15,16].
To probe the charge transfer and density functional theory (DFT), analysis can be employed to determine the nature of two molecular segments (i.e., molecule) in complexes based on their electronic structures, as illustrated in Figure 1. Donor–acceptor interaction energy calculation within the DFT framework plays a crucial role in studying charge transfer behaviours inside a molecular system. DFT analyses have been widely used to investigate CT complexes [17,18,19].
In the present research, we explore the electronic properties of three donor–acceptor complexes. The major investigation here is dedicated to the analysis of three distinct methodologies including the Mulliken population [20], Hirshfeld [21], and Voronoi [22]. These methods were used to trace down the charge transfer between the molecular segments (see Figure 1). CT calculations were first performed in isolated systems (i.e., gas phase), and then on a Au (111) surface (see Section S3 of the SI).
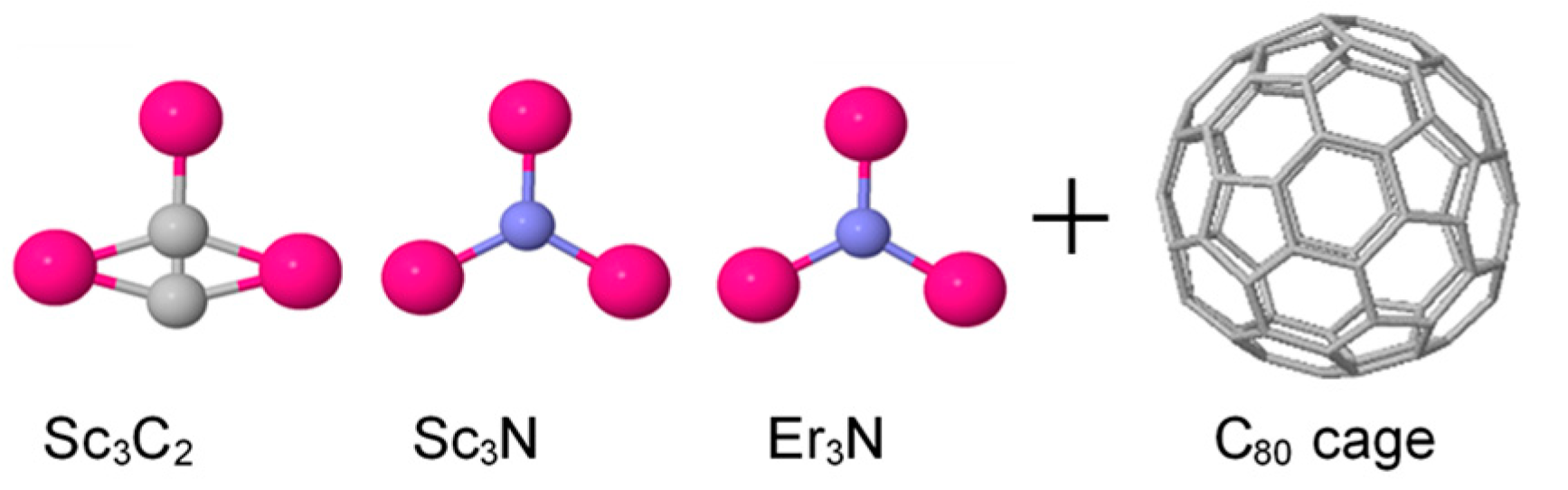
Figure 2 below illustrates the anatomy of three complexes, each of which consists of two molecular segments involving a metallic moiety such as Sc3C2, Sc3N, Er3N, and Ih-C80 cages. When the metallic moiety is encapsulated inside the fullerene cage, the outcome is endohedral metallofullerene (EMF) complexes. The current research investigates the electronic structure of three EMFs, i.e., Sc3C2@C80, Sc3N@C80, and Er3N@C80; examples of three EMF complexes and an empty fullerene are shown in Supplementary Figure S1 and Figure S2.
2. Computational Methods
All the theoretical simulations were carried out by employing the density functional (DFT) code SIESTA [23]. The optimum geometries of isolated EMFs were obtained by relaxing the molecules until all forces on the atoms were less than 0.01 eV/Å (for more detail, see Supplementary Figure S1 and Figure S2). A double-zeta plus polarization orbital basis set was used, with norm-conserving pseudopotentials and the local density approximation (LDA) exchange with a functional correlation, and to define the real space grid, an energy cut-off of 250 Rydberg was used. Results using GGA were also calculated and found that the resulting functions were comparable [24,25], with results obtained employing LDA functional exchange (see Section 1). To simulate the likely contact configuration during a break-junction experiment, we employed leads constructed from 6 layers of Au (111), each containing 30 gold atoms and further terminated with a pyramid of gold atoms.
To determine the optimum distance of EMF complexes attaching to the Au (111) metals, density functional theory and the counterpoise method were used, which removes basis set superposition errors (BSSEs). The binding distance was defined as the distance between the gold surface and the EMF complex. The ground state energy of the total system was calculated using SIESTA and is denoted . The energy of each monomer was then calculated in a fixed basis, which is achieved through the use of ghost atoms in SIESTA (see Section 4). Since the energy of the individual complex in the presence of the fixed basis is defined as and for the isolated gold as , the binding energy , is then calculated using the following equation [26,27,28]:
3. Results and Discussion
The electronic properties of the three EMF complexes involving Sc3C2@C80, Sc3N@C80, and Er3N@C80 were simulated employing both density functional theory (DFT) and quantum transport theory. To have a deep understanding of thermoelectric properties, the wave function of the investigated complexes, i.e., the lowest unoccupied orbitals (LUMO) and the highest occupied molecular orbitals (HOMO), along with their energies, are explored, as illustrated in Supplementary Figures S3–S5. These isosurface plots clearly demonstrate a significant weight on metallic moieties Sc3C2, Sc3N, and Er3N, in contrast to Ih-C80 cages. The significant weight occurs on the LUMO orbitals and it is well-known that these complexes possess LUMO dominated transport. This denotes that metallic moieties play a pivotal role in tuning the electronic properties of the EMF complexes.
As a first step, we investigated the charge transfer through these EMF complexes. Charge calculations are common practise in chemical science measurements and calculations. We shall first discuss charge transfer analyses in the gas phase for the three EMFs. We evaluate the net charge transfer from metallic moieties Sc3C2, Sc3N, and Er3N to Ih-C80 cages using three different DFT analyses methods Mulliken, Hirshfeld, and Voronoi (see Section S3 in the SI).
Table 1 below demonstrates that the three metallic moieties donate electrons to the C80 cage. However, the total number of the transferred electrons depends on the chemical nature (i.e., atom species) and geometrical shape of the metallic moiety. We find that the donation of erbium nitride Er3N is the highest followed by scandium nitride Sc3N and then scandium carbide Sc3C2. Furthermore, the charge transfer through the three EMF complexes follows the order Er3N@C80 > Sc3N@C80 > Sc3C2@C80 for Mulliken, Hirshfeld, and Voronoi analyses.
Surprisingly, there is a difference between the total number of the donated and gained electrons through the complexation (i.e., encapsulating the moiety inside cage). For example, the scandium carbide Sc3C2 donates 1.40 electrons to the cage; however, only 1.14 is indeed gained by the cage (loss–gain difference), and this occurs through all the EMF complexes. To answer this question, we tracked down the CT from the donor to receptor, atom by atom. The tracking analyses suggest the missing electrons are gained by the metallic moiety itself.
To accommodate this, we find that loss–gain differences are indeed gained by the moieties. For instance, through Sc3C2@C80 complexation, the two carbon atoms of Sc3C2, gain 0.26, 0.32, and 0.34 electrons from the moiety’s donation. Similarly, the nitrogen atoms of Sc3N and Er3N gain 0.24, 0.33, and 0.31 and 1.82, 1.34, and 1.32 electrons, respectively, when they form complexes with Ih-C80 cages (above analyses evaluated via the Mulliken population, Hirshfeld, and Voronoi). It should be noted that the electron travelled from the moiety to the Ih-C80 cage has a significant effect on the conductance and thermopower ; more details have been given previously [20,29]. Moreover, it should be noted that the total number of electrons donated by erbium nitride Er3N is significantly larger than of Sc3C2 and Sc3N moieties; we will discuss that later.
To mimic the likely metal–organic contact configuration during a scanning tunnelling microscope break-junction measurement (STM-BJ), we now repeat the above analysis for the case when the EMF forms a complex on a gold surface. Calculations in this section are for three parameters: metallic moiety, Au surface (2nd, 4th, and 6th columns), and a Ih-C80 cage (3rd, 5th, and 7th columns). Table 2 suggests that the electronic charge travels from both the metallic moiety and the gold surface to the Ih-C80 cage for the three configurations (i.e., EMF complex + Au surface). This behaviour is expected to occur as both the moiety and Au are metals, unlike the cage.
Table 2 explains that both the Sc3C2 and Au lose (+) electrons, and in total, their donation is 1.63 electrons (Sc3C2 = +1.33 and Au = +0.3 electrons). Again, only 1.39 is the gained (−) by the Ih-C80 cage, the difference of 0.24 electrons gained by C2 atoms within the moiety. Summing up the two negative figures (1.39 and 0.24), we obtain the total transferred electrons to be 1.63 electrons. Looking at the numbers in brackets mainly Au donation and C2, N gaining, one could summarise that Au donation is approximately 0.2–0.3 and C2 and N gaining 0.24–1.60 electrons. Furthermore, in all cases, the gain by C2 and N is larger than Au donation; we attribute that to the fact that C2 and N atoms are in direct contact with EMF moieties.
Again, in the erbium nitride configuration (Sc3N@C80 + Au), net charge transfers are significantly larger than those of the scandium nitride and scandium carbide configurations. Table 1 and Table 2 show that the net charge transfers of Er3N are more than four times higher than those of Sc3C2 and Sc3N, and the reason for this is that the erbium nitride moiety possesses f-electrons in its outer orbital shells [30]. It is widely known that DFT cannot treat electrons in f-orbitals accurately [31].
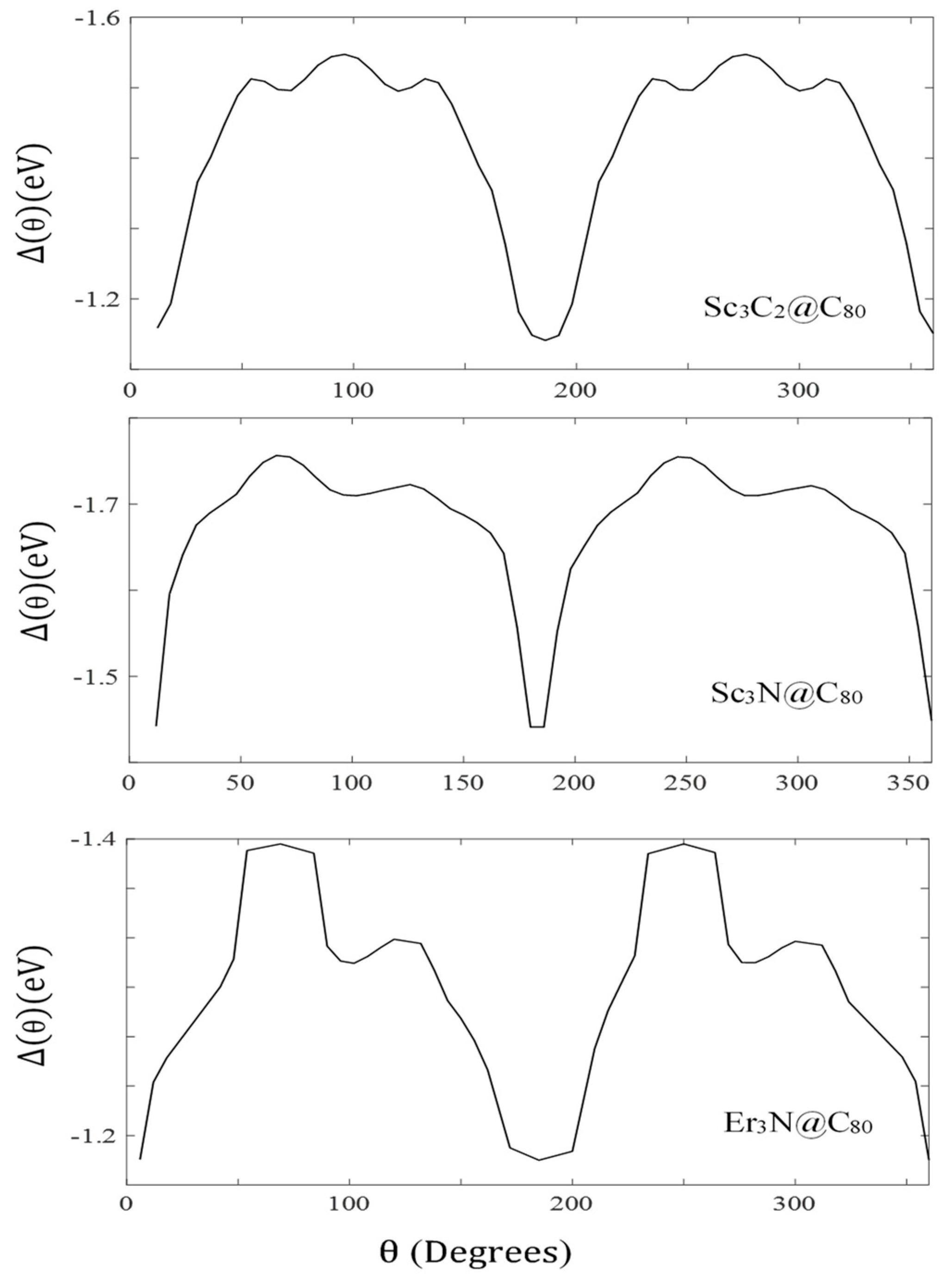
The above result explains why the of Er3N is less symmetric than that of Sc3C2 and Sc3N, as shown in Figure 3 (Note: Figure 3 is reported in our previous work [29]). This also applies to the charge inhomogeneity (). In ref. [29], the standard deviations of charge distributions on the three EMFs complexes were evaluated, and the values indicated that the of the erbium nitride complex was 10 times less than that of the other complexes, as illustrated in Table 3.
CT analyses that were performed in both the solute and on a Au surface are essential to determine the total number of electrons transferred from metallic moieties to the Ih-C80 cage. These charge transfers have a great influence on the electric and thermoelectric properties of the EMFs. The electrical conductance is boosted in crown ether molecules [32,33], due to charge transfer from the ion to the molecular wire, causing the molecular resonances to shift closer to the electrode Fermi energy. Similarly, CT enhances the Seebeck coefficient in crown ether molecules [34,35,36,37] and endohedral metallofullerenes [38,39,40].
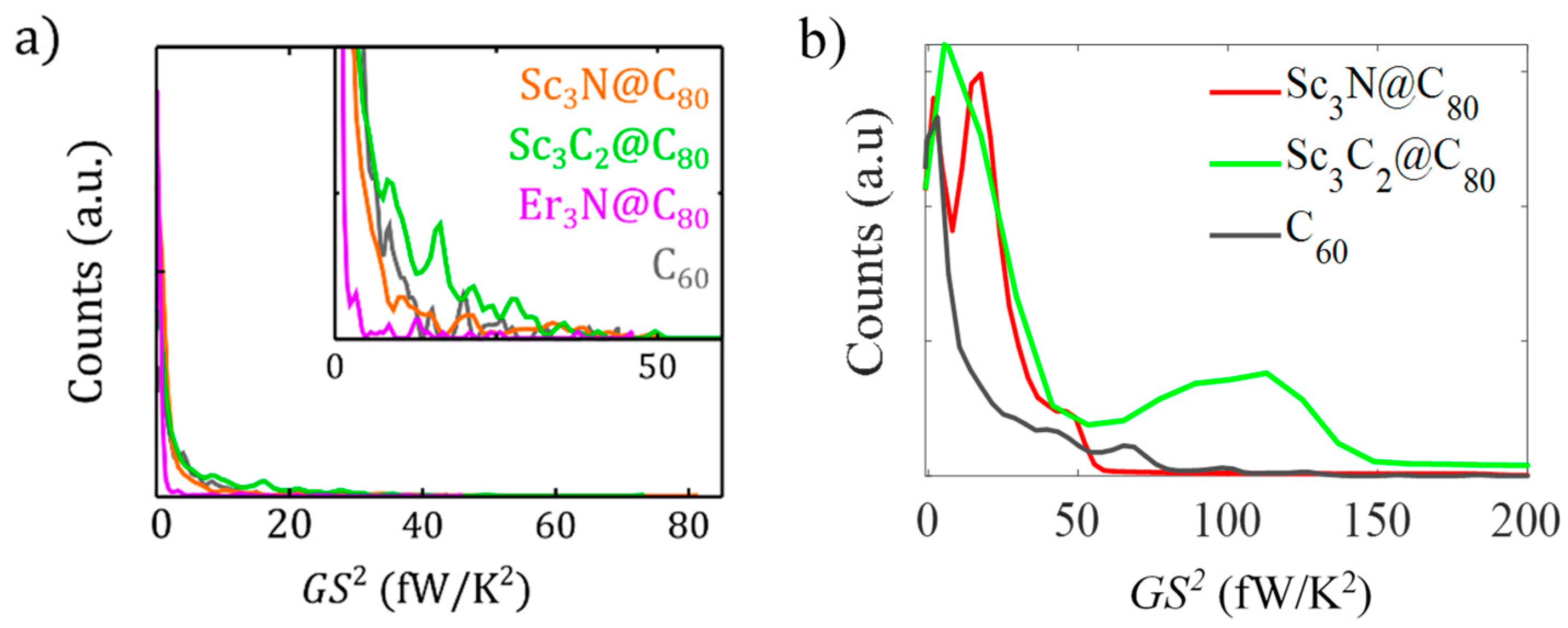
Many experimental and theoretical studies pointed out that EMF–complex junctions retain a high single molecule power factor. For example, Lee and his co-workers [41] reported that the Gd@C82 complex has the biggest power factor for a molecular device (at the time of publication), which is about 16.2 fW K−2. This is equivalent to approximately 4 × 10 μW K−2 m−1 for a Gd@C82 monolayer. In another study [29] performed in 2022, the researchers noticed a larger PF of 50 fW K−2 for Sc3N@C80 and Sc3C2@C80 complexes, and some measurements hit 70–80 fW K−2 for Sc3N@C80 and Sc3C2@C80 complexes. Statistically, they report larger values for the carbide complex (Sc3C2@C80). Considering all their measured conductance and Seebeck coefficient values, the PF can be statistically improved when the charge transfer becomes larger. We attribute this desirable feature to the CT phenomena. Table 3 above clearly illustrates that the charge transfer of Sc3N@C80 and Sc3C2@C80 complexes are approximately 10 times larger than that of the Er3N@C80 complex, and this explains why their conductance and Seebeck (i.e., power factor ) are larger than those of Er3N@C80, as shown in Figure 4 below (Note: Figure 4 has been reported in our previous work [29]).
Large power factor and bi-thermoelectric behaviour of the studied EMF complexes underline our initial intuition that charge transfer (CT) results in considerable improvement in the thermoelectric transport properties, compared with an empty cage such as C60. This desirable feature has also been noted previously [41] in some EMF complexes involving Gd@C82 and Ce@C82, and the empty C82 displayed mainly negative Seebeck coefficients, with occasional positive Seebeck coefficients. The positive and negative Seebeck coefficients were ascribed to meta-geometries. The reported findings of the thermopower demonstrate improvements in the EMFs thermoelectric properties compared to the empty C82. Compared to the current investigated EMF complexes, the only difference with the complexes explored in a previous study [41] is the number of metallic atoms within the complex, and in [41], a single atom was positioned out within the cage. This difference could lead only to less migrated charge to the fullerene cage as the metallic moiety is smaller.
4. Conclusions
In conclusion, through a systematic theory study, we have demonstrated that the electrical and thermoelectrical performance of endohedral metallofullerene (EMF) complexes and configurations can be modulated by chemically varying the metallic moiety that encapsulates inside Ih-C80 cages. The electric charge transfer of three EMFs involving Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes and their configurations when they are placed on a gold (111) surface have been investigated in three different charge transfer methods. The Mulliken, Hirshfeld, and Voronoi methods all suggest that the charge migrates from metallic moieties such as Sc3C2, Sc3N, and Er3N to C80 cages; however, the amount of the transferred charge depends on the nature of the moiety inside the EMF complex. Published studies [32,33,38,42,43,44,45] evidenced that the CT improve both conductance and thermopower. This work sheds light on new strategies for designing electric and thermoelectric devices based on tuning the CT by using different metallic moieties with potential practical applications.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/en16114342/s1, Figure S1: Geometries of an asymmetric Sc3C2 (a), symmetric Sc3N and Er3N moieties (b,c). Key: C = grey, N = blue and Er = O = red; Figure S2: Endohedral metallofullerenes and fullerene studied Molecules. Schematic of the three endohedral metallofullerenes (EMFs), namely, a: Sc3C2@C80, b: Sc3N@C80, and c: Er3N@C80 and an empty fullerene cage d: C80; Figure S3: Wave function plots of Sc3C2@C80 complex. Top panel: fully optimised geometry of Sc3C2@C80 EMF. Lower panel: HOMO, LUMO, HOMO-1, LUMO+1 of Sc3C2@C80 complex along with their energies; Figure S4: Wave function plots of Er3N@C80 complex. Top panel: fully optimised geometry of Sc3C2@C80 EMF. Lower panel: HOMO, LUMO, HOMO-1, LUMO+1 of Er3N@C80 complex along with their energies; Figure S5: Wave function plots of Er3N@C80 complex. Top panel: fully optimised geometry of Sc3C2@C80 EMF. Lower panel: HOMO, LUMO, HOMO-1, LUMO+1 of Er3N@C80 complex along with their energies; Figure S6: Sc3C2@C80 on a gold surface (Right panel). Energy difference of Sc3C2@C80 /gold complex as a function of molecule-gold distance. The equilibrium distance corresponding to the energy minimum is found to be approximately 2.5\Å (Left panel); Figure S7: Seebeck coefficient S as a function of Fermi energy at 60 different orientations angles \theta of Sc3C2@C80, for a tip-substrate distance of 2.5 Å; Figure S8: Seebeck coefficients S as a function of Fermi energy at 60 different orientation angles\theta of Sc3N@C80 for a tip-substrate distance of 2.5 Å. Table S1: Charge transfer analyses using Mulliken, Hirshfeld and Voronoi methods of Sc3C2@C80, Sc3N@C80 and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties (with a charge of +|e|), to Ih-C80 cages (with a charge of −|e|), to form complexes. Note: loss-gain differences gain by C2, N and N (numbers in brackets), Sc3C2@C80, Sc3N@C80 and Er3N@C80 complexes in gas phase; Table S2: Charge transfer analyses using Mulliken, Hirshfeld and Voronoi methods of Sc3C2@C80, Sc3N@C80 and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties (with a charge of +|e|), to Ih-C80 cages (with a charge of −|e|), to form complexes. Note: loss-gain differences gain by C2, N and N (numbers in brackets), of Sc3C2@C80, Sc3N@C80 and Er3N@C80 complexes on an Au (111), surface.
Author Contributions
A.K.I. originally conceived the concept; calculations were carried out by M.A. (Majed Alshammari), M.A. (Moteb Altoaibi) and T.A. All authors provided essential contributions to interpreting the data reported in this manuscript. A.K.I. coordinated the writing of the manuscript with input from M.A. (Majed Alshammari), M.A. (Moteb Altoaibi) and T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Leverhulme Trust for Early Career Fellowship ECF-2020-638. This work was additionally funded by the European Commission FET Open projects 767187-QuIET and 766853-EFINED. M.A. (Majed Alshammari) and T.A. are grateful for the financial assistance from Jouf University (Saudi Arabia), M.A. (Majed Alshammari) and T.A. are thankful for computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia. M.A. (Moteb Altoaibi) is grateful for the sported the Deanship of Scientific Research at Prince Sattam bin Abdulaziz University, Alkharj, SaudiArabia and the Saudi Ministry of Education. A.K.I. is grateful for financial assistance from Tikrit University (Iraq), and the Iraqi Ministry of Higher Education (SL-20).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soos, Z.G. Theory of π-molecular charge-transfer crystals. Annu. Rev. Phys. Chem. 1974, 25, 121–153. [Google Scholar] [CrossRef]
- Bauer, C.; Teuscher, J.; Brauer, J.C.; Punzi, A.; Marchioro, A.; Ghadiri, E.; De Jonghe, J.; Wielopolski, M.; Banerji, N.; Moser, J.-E. Dynamics and mechanisms of interfacial photoinduced electron transfer processes of third generation photovoltaics and photocatalysis. CHIMIA Int. J. Chem. 2011, 65, 704–709. [Google Scholar] [CrossRef]
- Günes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated polymer-based organic solar cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Méndez-Hernández, D.D.; Tejeda-Ferrari, M.E.; Teillout, A.-L.; Llansola-Portolés, M.J.; Kodis, G.; Poluektov, O.G.; Rajh, T.; Mujica, V.; Groy, T.L. A bioinspired redox relay that mimics radical interactions of the Tyr–His pairs of photosystem. Nat. Chem. 2014, 6, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Bull. Am. Phys. Soc. 1974, 19, 341. [Google Scholar] [CrossRef]
- Herrer, L.; Ismael, A.; Martin, S.; Milan, D.C.; Serrano, J.L.; Nichols, R.J.; Lambert, C.; Cea, P. Single molecule vs. large area design of molecular electronic devices incorporating an efficient 2-aminepyridine double anchoring group. Nanoscale 2019, 11, 15871–15880. [Google Scholar] [CrossRef]
- Al-Khaykanee, M.K.; Ismael, A.K.; Grace, I.; Lambert, C.J. Oscillating Seebeck coefficients in π-stacked molecular junctions. Rsc Adv. 2018, 8, 24711–24715. [Google Scholar] [CrossRef]
- Bockrath, M.; Cobden, D.H.; McEuen, P.L.; Chopra, N.G.; Zettl, A.; Thess, A.; Smalley, R.E. Single-electron transport in ropes of carbon nanotubes. Science 1997, 275, 1922–1925. [Google Scholar] [CrossRef]
- Ismael, A.K.; Lambert, C.J. Single-molecule conductance oscillations in alkane rings. J. Mater. Chem. C 2019, 7, 6578–6581. [Google Scholar] [CrossRef]
- Romaner, L.; Heimel, G.; Brédas, J.-L.; Gerlach, A.; Schreiber, F.; Johnson, R.L.; Zegenhagen, J.; Duhm, S.; Koch, N.; Zojer, E. Impact of bidirectional charge transfer and molecular distortions on the electronic structure of a metal-organic interface. Phys. Rev. Lett. 2007, 99, 256801. [Google Scholar] [CrossRef]
- Bennett, T.L.; Alshammari, M.; Au-Yong, S.; Almutlg, A.; Wang, X.; Wilkinson, L.A.; Albrecht, T.; Jarvis, S.P.; Cohen, L.F.; Ismael, A. Multi-component self-assembled molecular-electronic films: Towards new high-performance thermoelectric systems. Chem. Sci. 2022, 13, 5176–5185. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Chen, G.; Perry, J.W.; Goddard, W.A., III. Valence-bond charge-transfer model for nonlinear optical properties of charge-transfer organic molecules. J. Am. Chem. Soc. 1994, 116, 10679–10685. [Google Scholar] [CrossRef]
- Gorczak, N.; Renaud, N.; Tarkuç, S.; Houtepen, A.J.; Eelkema, R.; Siebbeles, L.D.; Grozema, F.C. Charge transfer versus molecular conductance: Molecular orbital symmetry turns quantum interference rules upside down. Chem. Sci. 2015, 6, 4196–4206. [Google Scholar] [CrossRef] [PubMed]
- Closs, G.L.; Miller, J.R. Intramolecular long-distance electron transfer in organic molecules. Science 1988, 240, 440–447. [Google Scholar] [CrossRef]
- Sukegawa, J.; Schubert, C.; Zhu, X.; Tsuji, H.; Guldi, D.M.; Nakamura, E. Electron transfer through rigid organic molecular wires enhanced by electronic and electron–vibration coupling. Nat. Chem. 2014, 6, 899–905. [Google Scholar] [CrossRef]
- Deibel, C.; Strobel, T.; Dyakonov, V. Role of the charge transfer state in organic donor–acceptor solar cells. Adv. Mater. 2010, 22, 4097–4111. [Google Scholar] [CrossRef]
- Otero, R.; de Parga, A.V.; Gallego, J.M. Electronic, structural and chemical effects of charge-transfer at organic/inorganic interfaces. Surf. Sci. Rep. 2017, 72, 105–145. [Google Scholar] [CrossRef]
- Kollmannsberger, M.; Rurack, K.; Resch-Genger, U.; Rettig, W.; Daub, J. Design of an efficient charge-transfer processing molecular system containing a weak electron donor: Spectroscopic and redox properties and cation-induced fluorescence enhancement. Chem. Phys. Lett. 2000, 329, 363–369. [Google Scholar] [CrossRef]
- Wörner, H.J.; Arrell, C.A.; Banerji, N.; Cannizzo, A.; Chergui, M.; Das, A.K.; Hamm, P.; Keller, U.; Kraus, P.M.; Liberatore, E. Charge migration and charge transfer in molecular systems. Struct. Dyn. 2017, 4, 061508. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Guerra, C.F.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D.J.J.o.P.C.M. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Davidson, R.J.; Milan, D.C.; Al-Owaedi, O.A.; Ismael, A.K.; Nichols, R.J.; Higgins, S.J.; Lambert, C.J.; Yufit, D.S.; Beeby, A. Conductance of ‘bare-bones’ tripodal molecular wires. RSC Adv. 2018, 8, 23585–23590. [Google Scholar] [CrossRef] [PubMed]
- Markin, A.; Ismael, A.K.; Davidson, R.J.; Milan, D.C.; Nichols, R.J.; Higgins, S.J.; Lambert, C.J.; Hsu, Y.-T.; Yufit, D.S.; Beeby, A. Conductance Behavior of Tetraphenyl-Aza-BODIPYs. J. Phys. Chem. C 2020, 124, 6479–6485. [Google Scholar] [CrossRef]
- Kobko, N.; Dannenberg, J. Dannenberg. Effect of basis set superposition error (BSSE) upon ab initio calculations of organic transition states. J. Phys. Chem. A 2001, 105, 1944–1950. [Google Scholar] [CrossRef]
- Sherrill, C.D. Counterpoise Correction and Basis Set Superposition Error; School of Chemistry and Biochemistry, Georgia Institute of Technology: Atlanta, Georgia, 2010. [Google Scholar]
- Sinnokrot, M.O.; Valeev, E.F.; Sherrill, C.D. Estimates of the ab initio limit for π− π interactions: The benzene dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. [Google Scholar] [CrossRef]
- Ismael, A.K.; Rincón-García, L.; Evangeli, C.; Dallas, P.; Alotaibi, T.; Al-Jobory, A.A.; Rubio-Bollinger, G.; Porfyrakis, K.; Agraït, N.; Lambert, C.J. Exploring seebeck-coefficient fluctuations in endohedral-fullerene, single-molecule junctions. Nanoscale Horiz. 2022, 7, 616–625. [Google Scholar] [CrossRef]
- Akkermans, E.; Montambaux, G. Mesoscopic Physics of Electrons and Photons; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Ismael, A.K.; Al-Jobory, A.; Grace, I.; Lambert, C.J. Discriminating single-molecule sensing by crown-ether-based molecular junctions. J. Chem. Phys. 2017, 146, 064704. [Google Scholar] [CrossRef]
- Ismael, A.K.; Grace, I.; Lambert, C.J. Increasing the thermopower of crown-ether-bridged anthraquinones. Nanoscale 2015, 7, 17338–17342. [Google Scholar] [CrossRef] [PubMed]
- Ismael, A.K.; Grace, I.; Lambert, C.J. Connectivity dependence of Fano resonances in single molecules. Phys. Chem. Chem. Phys. 2017, 19, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ismael, A.; Ning, S.; Althobaiti, H.; Al-Jobory, A.; Girovsky, J.; Astier, H.P.; O'Driscoll, L.J.; Bryce, M.R.; Lambert, C.J. Electrostatic Fermi level tuning in large-scale self-assembled monolayers of oligo (phenylene–ethynylene) derivatives. Nanoscale Horiz. 2022, 7, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L.A.; Bennett, T.L.; Grace, I.M.; Hamill, J.; Wang, X.; Au-Yong, S.; Ismael, A.; Jarvis, S.P.; Hou, S.; Albrecht, T. Assembly, structure and thermoelectric properties of 1,1′-dialkynylferrocene ‘hinges’. Chem. Sci. 2022, 13, 8380–8387. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Al-Jobory, A.; Zhang, Q.-C.; Cao, W.; Alshehab, A.; Qu, K.; Alotaibi, T.; Chen, H.; Liu, J.; Ismael, A.K. Highly insulating alkane rings with destructive σ-interference. Sci. China Chem. 2022, 65, 1822–1828. [Google Scholar] [CrossRef]
- Rincón-García, L.; Ismael, A.K.; Evangeli, C.; Grace, I.; Rubio-Bollinger, G.; Porfyrakis, K.; Agraït, N.; Lambert, C.J. Molecular design and control of fullerene-based bi-thermoelectric materials. Nat. Mater. 2016, 15, 289–293. [Google Scholar] [CrossRef]
- Lu, J.; Nagase, S.; Zhang, X.; Wang, D.; Ni, M.; Maeda, Y.; Wakahara, T.; Nakahodo, T.; Tsuchiya, T.; Akasaka, T. Selective interaction of large or charge-transfer aromatic molecules with metallic single-wall carbon nanotubes: Critical role of the molecular size and orientation. J. Am. Chem. Soc. 2006, 128, 5114–5118. [Google Scholar] [CrossRef]
- Ismael, A.; Al-Jobory, A.; Wang, X.; Alshehab, A.; Almutlg, A.; Alshammari, M.; Grace, I.; Benett, T.L.; Wilkinson, L.A.; Robinson, B.J. Molecular-scale thermoelectricity: As simple as ‘ABC’. Nanoscale Adv. 2020, 2, 5329–5334. [Google Scholar] [CrossRef]
- Lee, S.K.; Buerkle, M.; Yamada, R.; Asai, Y.; Tada, H. Thermoelectricity at the molecular scale: A large Seebeck effect in endohedral metallofullerenes. Nanoscale 2015, 7, 20497–20502. [Google Scholar] [CrossRef]
- Balachandran, J.; Reddy, P.; Dunietz, B.D.; Gavini, V. End-group-induced charge transfer in molecular junctions: Effect on electronic-structure and thermopower. J. Phys. Chem. Lett. 2012, 3, 1962–1967. [Google Scholar] [CrossRef]
- Adams, D.M.; Brus, L.; Chidsey, C.E.; Creager, S.; Creutz, C.; Kagan, C.R.; Kamat, P.V.; Lieberman, M.; Lindsay, S.; Marcus, R.A. Charge transfer on the nanoscale: Current status. J. Phys. Chem. B 2003, 107, 6668–6697. [Google Scholar] [CrossRef]
- Liu, S.-X.; Ismael, A.K.; Al-Jobory, A.; Lambert, C.J. Signatures of Room-Temperature Quantum Interference in Molecular Junctions. Acc. Chem. Res. 2023, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Alshehab, A.; Ismael, A.K. Impact of the terminal end-group on the electrical conductance in alkane linear chains. RSC Adv. 2023, 13, 5869–5873. [Google Scholar] [CrossRef] [PubMed]
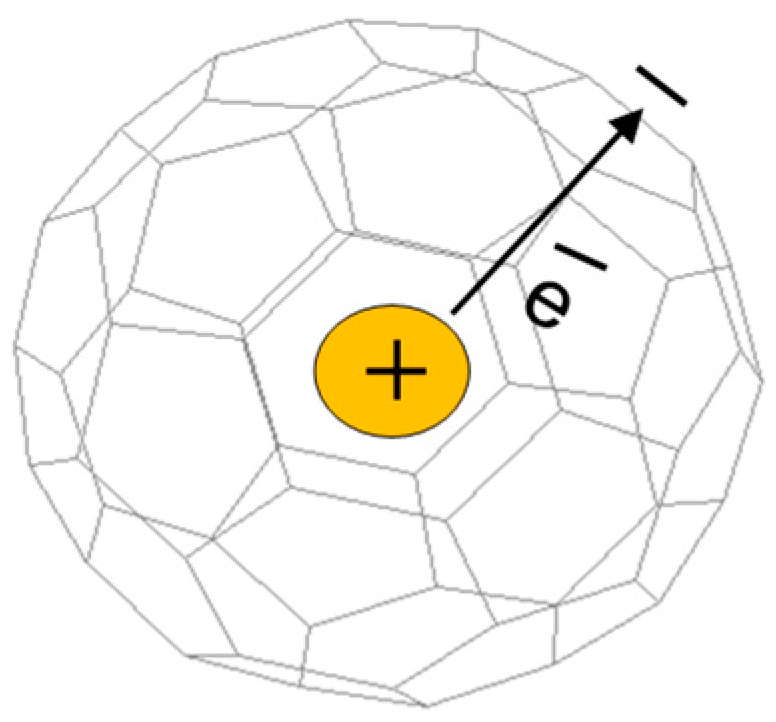
Figure 1.
Schematic illustration of donor–acceptor complex Li@C60. Li cation is positively charged (donor), while C60 cage is negatively charged (acceptor).
Figure 1.
Schematic illustration of donor–acceptor complex Li@C60. Li cation is positively charged (donor), while C60 cage is negatively charged (acceptor).

Figure 2.
Schematic illustration of three metallic moieties including Sc3C2, Sc3N, Er3N, and Ih-C80 cages (Note: All structures are fully optimised). Insertion of the metallic moiety inside the cage yields EMF complexes such as Sc3C2@C80, Sc3N@C80, and Er3N@C80.
Figure 2.
Schematic illustration of three metallic moieties including Sc3C2, Sc3N, Er3N, and Ih-C80 cages (Note: All structures are fully optimised). Insertion of the metallic moiety inside the cage yields EMF complexes such as Sc3C2@C80, Sc3N@C80, and Er3N@C80.

Figure 3.
of Er3N, Sc3C2, and Sc3N within the fullerene cage. Energy barriers to rotation about . Adapted with permission from [29]. Copyright 2022 Nanoscale Horizons, 2022.
Figure 3.
of Er3N, Sc3C2, and Sc3N within the fullerene cage. Energy barriers to rotation about . Adapted with permission from [29]. Copyright 2022 Nanoscale Horizons, 2022.

Figure 4.
analysis. (a) Experimental histograms of PF at first contact, built with the data in Figure 3. The inset zooms into the details of the main panel. (b). Theoretical 1D histograms of power factor obtained from Supplementary Figure S14 and Figure S15 of the Supplementary Materials. Adapted with permission from [29]. Copyright 2022 Nanoscale Horizons, 2022.
Figure 4.
analysis. (a) Experimental histograms of PF at first contact, built with the data in Figure 3. The inset zooms into the details of the main panel. (b). Theoretical 1D histograms of power factor obtained from Supplementary Figure S14 and Figure S15 of the Supplementary Materials. Adapted with permission from [29]. Copyright 2022 Nanoscale Horizons, 2022.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Gas phase, charge transfer calculations employing Mulliken, Hirshfeld, and Voronoi methods of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties (with a charge of +|e|) to Ih-C80 cages (with a charge of –|e|) to form complexes. Note: loss–gain differences gained by C2, N, and N (numbers in brackets) of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes.
Table 1.
Gas phase, charge transfer calculations employing Mulliken, Hirshfeld, and Voronoi methods of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties (with a charge of +|e|) to Ih-C80 cages (with a charge of –|e|) to form complexes. Note: loss–gain differences gained by C2, N, and N (numbers in brackets) of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes.
| Metallic Moiety | Mulliken | Hirshfeld | Voronoi | |||
|---|---|---|---|---|---|---|
| moiety | cage | moiety | cage | moiety | cage | |
| Sc3C2 | +1.40 | −1.14 | +1.15 | −0.83 | +1.06 | −0.72 |
| C2 | (−0.26) | - | (−0.32) | - | (−0.34) | - |
| Sc3N | +1.50 | −1.26 | +1.31 | −0.98 | +1.27 | −0.96 |
| N | (−0.24) | - | (−0.33) | - | (−0.31) | - |
| Er3N | +6.96 | −5.14 | +7.48 | −6.14 | +7.14 | −5.82 |
| N | (−1.82) | - | (−1.34) | - | (−1.32) | - |
Table 2.
On a gold surface, charge transfer calculations employing the Mulliken, Hirshfeld, and Voronoi methods of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties and Au surface (with a charge of +|e|) to Ih-C80 cages (with a charge of −|e|) to form complex Au junctions. Note: numbers in brackets correspond to Au donation (+|e|) and C2, N gaining (−|e|).
Table 2.
On a gold surface, charge transfer calculations employing the Mulliken, Hirshfeld, and Voronoi methods of Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. The total number of electrons transferred from metallic moieties and Au surface (with a charge of +|e|) to Ih-C80 cages (with a charge of −|e|) to form complex Au junctions. Note: numbers in brackets correspond to Au donation (+|e|) and C2, N gaining (−|e|).
| Moiety + Au | Mulliken | Hirshfeld | Voronoi | |||
|---|---|---|---|---|---|---|
| moiety | cage | moiety | cage | moiety | cage | |
| Sc3C2 | +1.33 | −1.39 | +0.93 | –0.81 | +0.97 | −0.79 |
| Au, C2 | (+0.3, −0.24) | - | (+0.2, −0.32) | - | (+0.18, −0.36) | - |
| Sc3N | +2.15 | −2.09 | +1.07 | –0.98 | +1.04 | –1.02 |
| Au, N | (+0.22, −0.28) | - | (+0.23, −0.32) | - | (+0.24, −0.26) | - |
| Er3N | +6.53 | −5.20 | +6.96 | –5.80 | +6.66 | −5.60 |
| Au, N | (+0.24, −1.57) | - | (+0.28, −1.44) | - | (+0.29, −1.35) | - |
Table 3.
Standard deviations of charge for Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. Charges are simulated using the Mulliken, Hirshfeld, and Voronoi methods. Adapted with permission from ref [29]. Copyright 2022 Nanoscale Horizons, 2022.
Table 3.
Standard deviations of charge for Sc3C2@C80, Sc3N@C80, and Er3N@C80 complexes. Charges are simulated using the Mulliken, Hirshfeld, and Voronoi methods. Adapted with permission from ref [29]. Copyright 2022 Nanoscale Horizons, 2022.
| EMF Complex | |||
|---|---|---|---|
| Sc3C2@C80 | 0.0154 | 0.0113 | 0.0133 |
| Sc3N@C80 | 0.0163 | 0.0109 | 0.0119 |
| Er3N@C80 | 0.00378 | 0.00259 | 0.00268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alshammari, M.; Alotaibi, T.; Alotaibi, M.; Ismael, A.K. Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes. Energies 2023, 16, 4342. https://0-doi-org.brum.beds.ac.uk/10.3390/en16114342
AMA Style
Alshammari M, Alotaibi T, Alotaibi M, Ismael AK. Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes. Energies. 2023; 16(11):4342. https://0-doi-org.brum.beds.ac.uk/10.3390/en16114342
Chicago/Turabian StyleAlshammari, Majed, Turki Alotaibi, Moteb Alotaibi, and Ali K. Ismael. 2023. "Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes" Energies 16, no. 11: 4342. https://0-doi-org.brum.beds.ac.uk/10.3390/en16114342
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.