Structure of Plasma (re)Polymerized Polylactic Acid Films Fabricated by Plasma-Assisted Vapour Thermal Deposition

 ,
,  ,
, 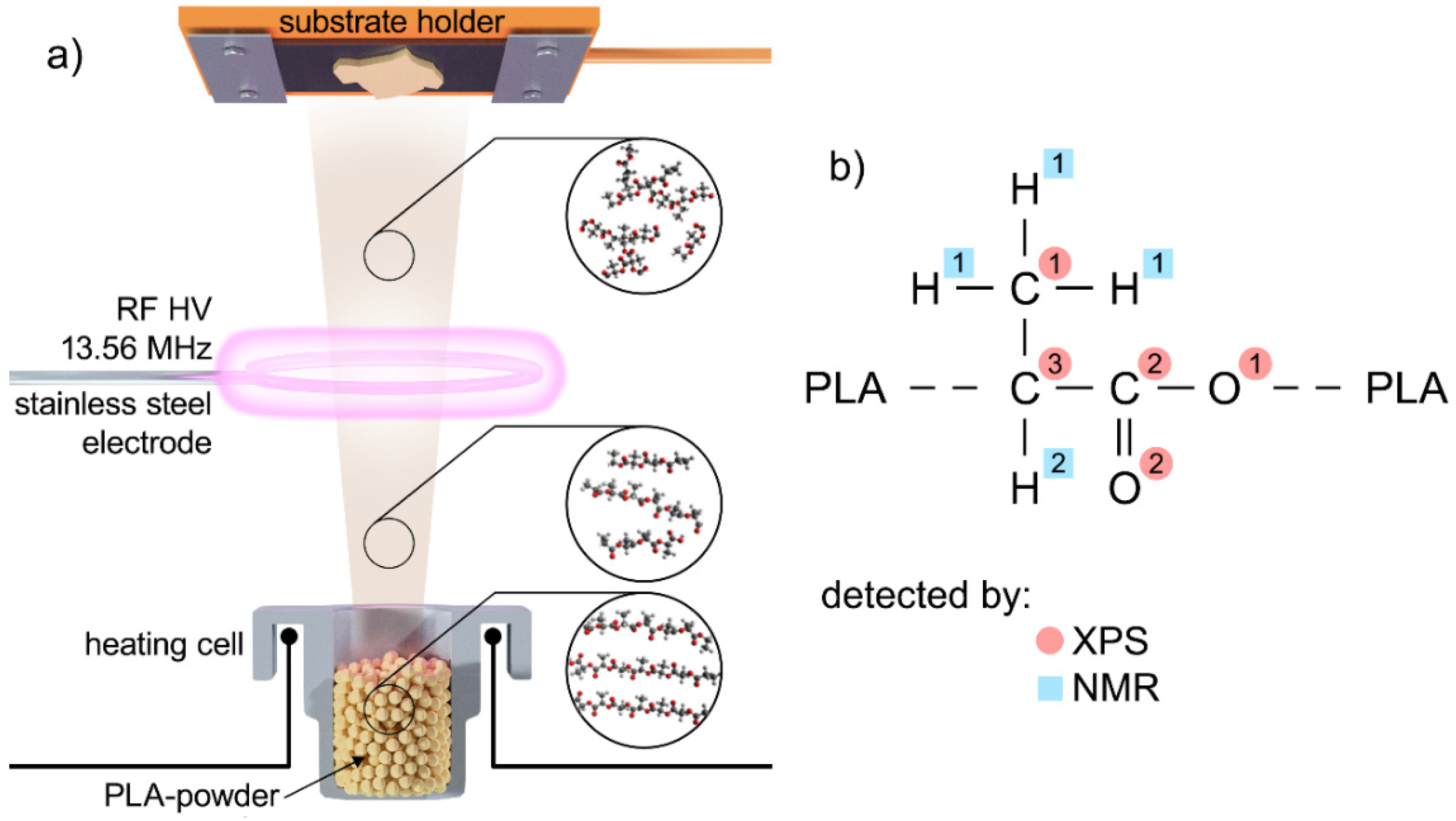{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Deposition of Plasma Polymerised Polylactic Acid (PLA) Coatings
2.2. Characterisation of Deposited Films
3. Results
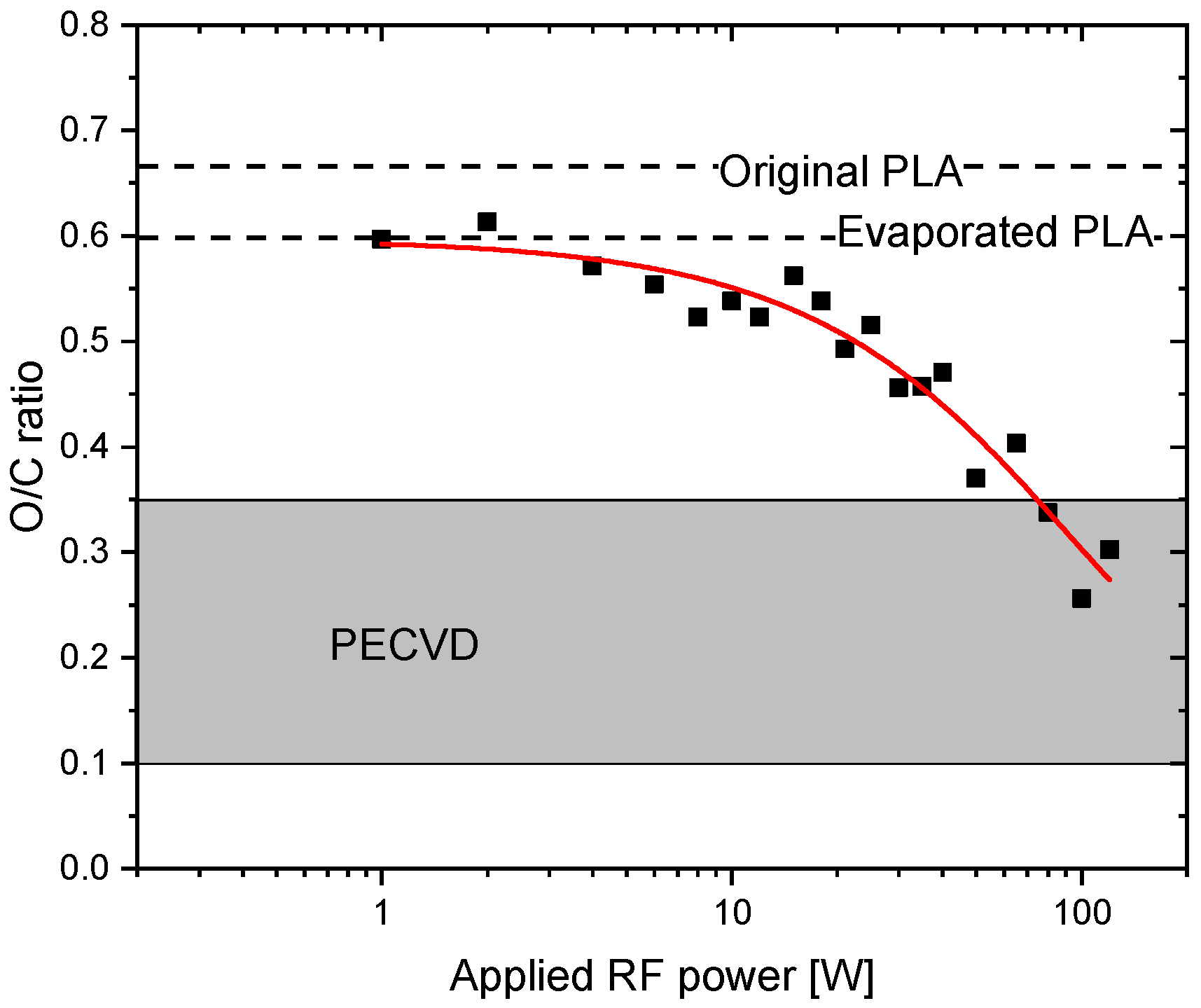
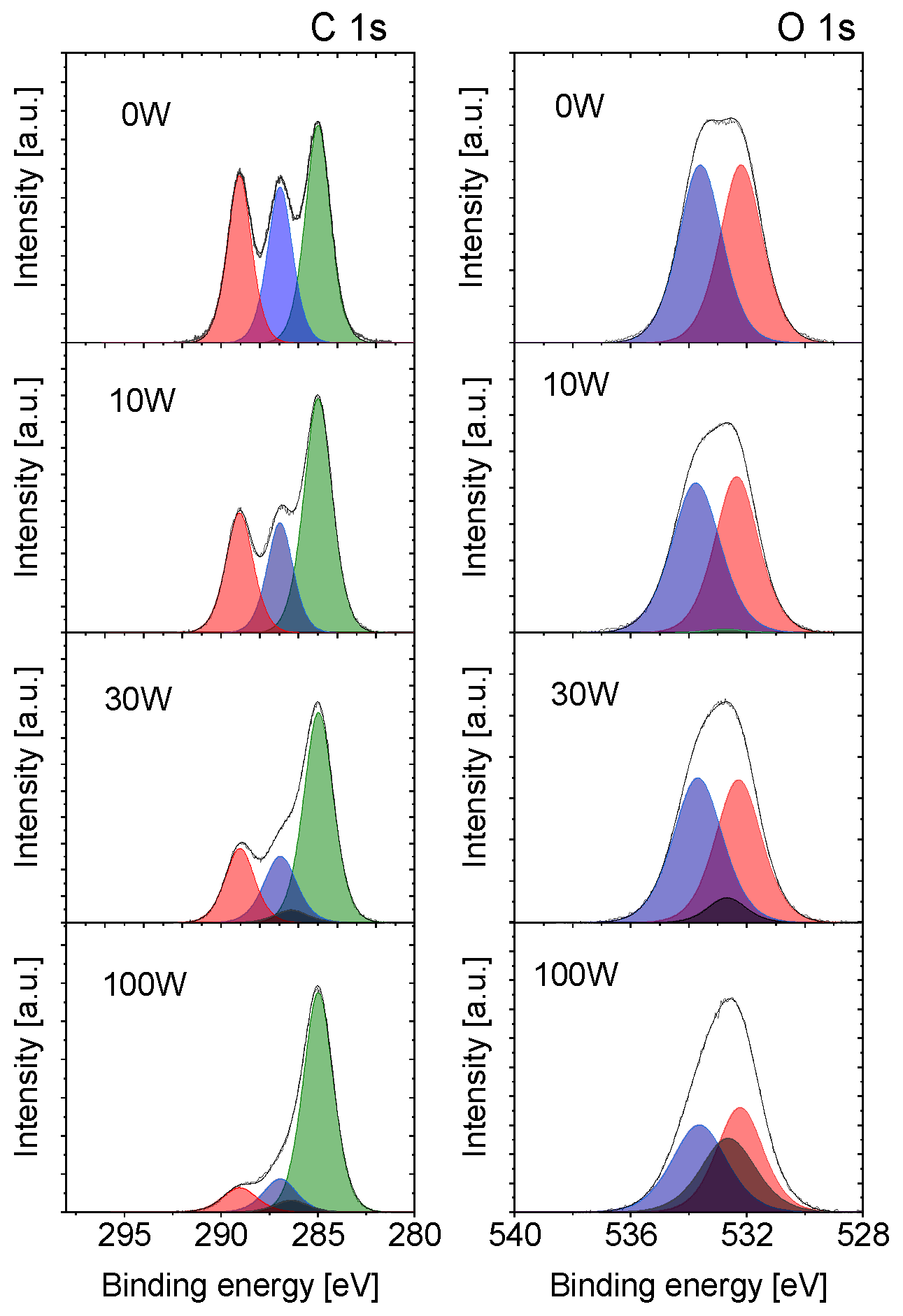
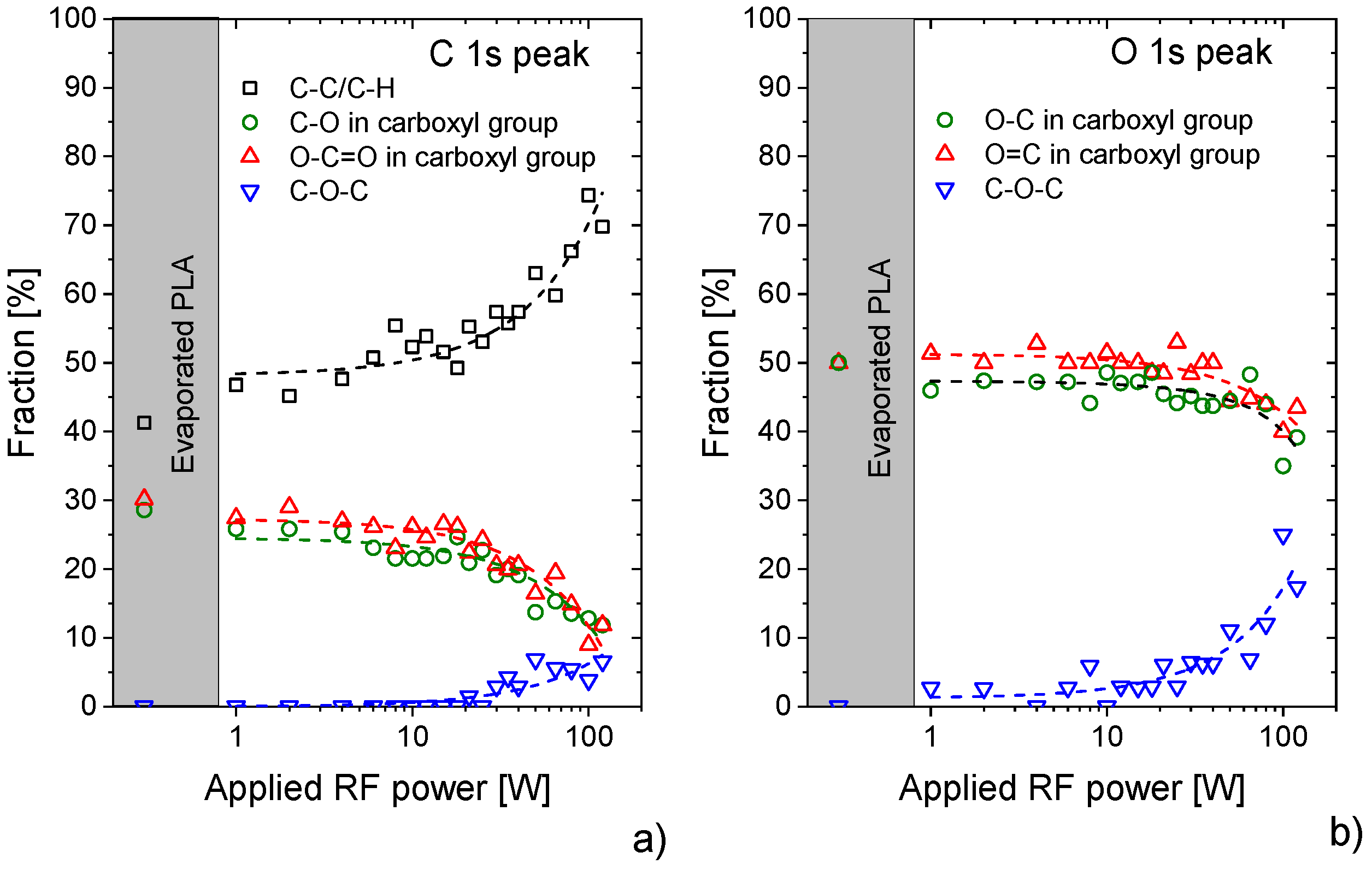
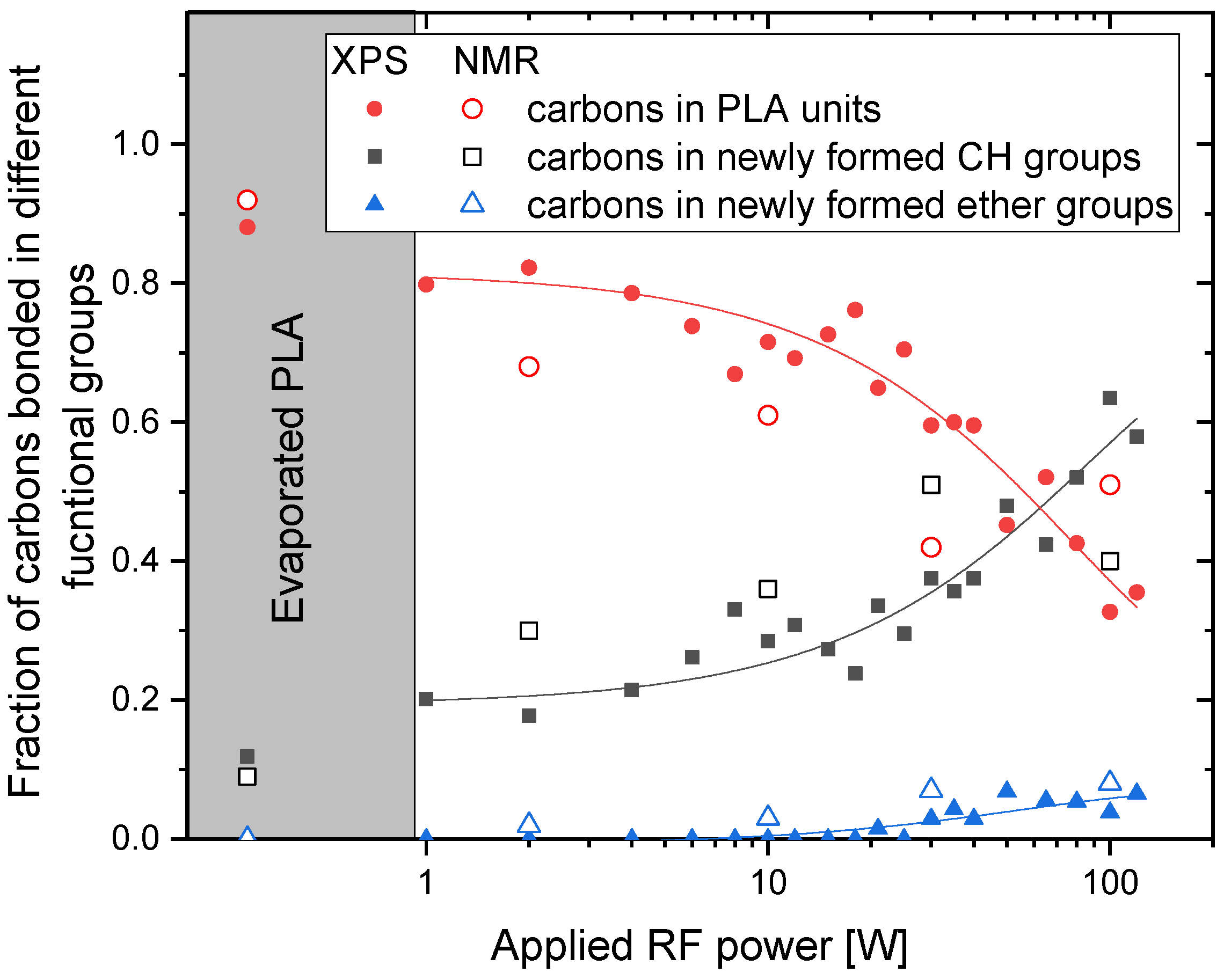
3.1. X-ray Photoelectron Spectroscopy (XPS) Characterisation of Plasma Polymerised PLA Coatings
3.2. NMR Analysis of PAVTD-Deposited PLA-Like Coatings
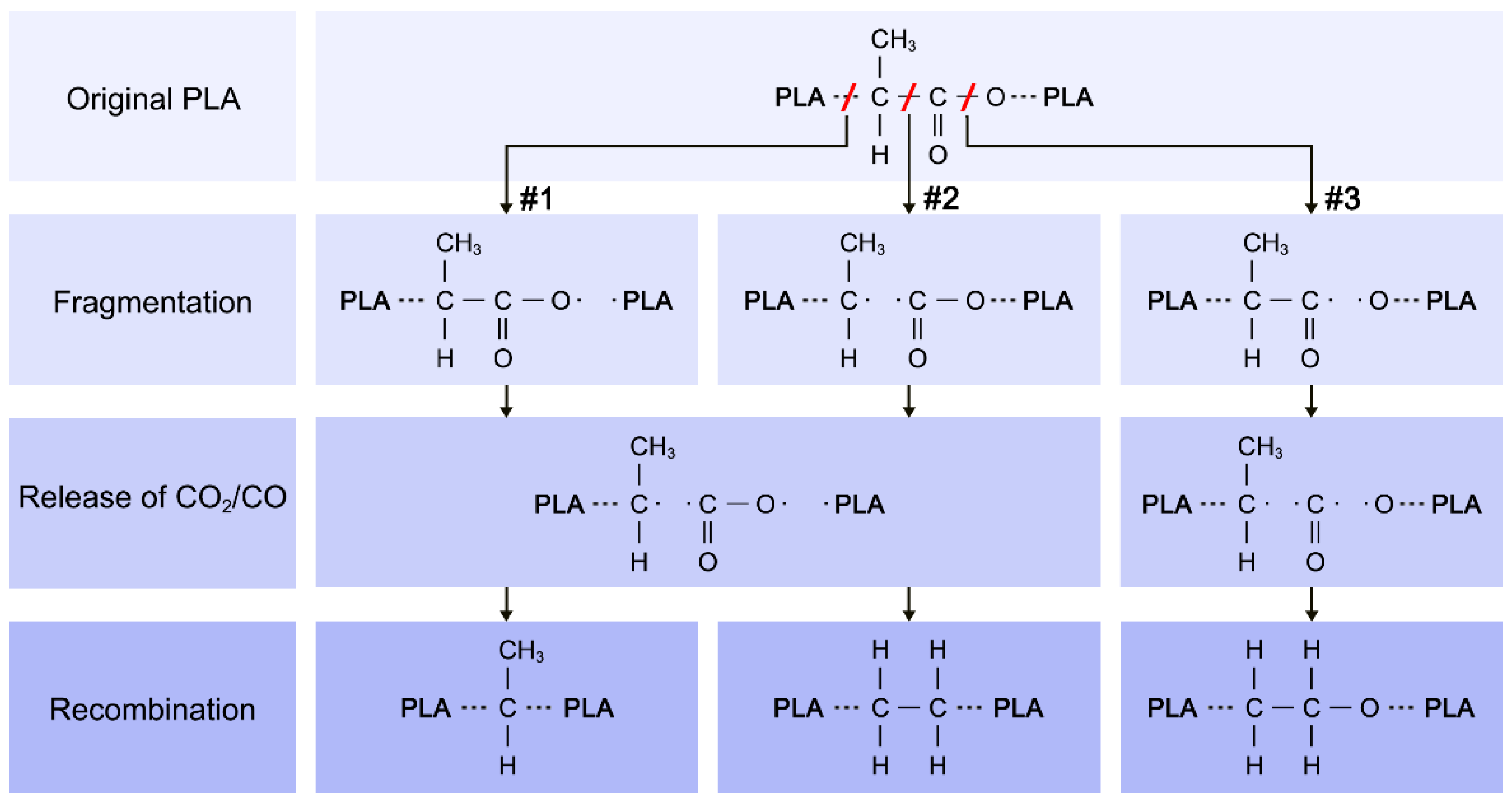
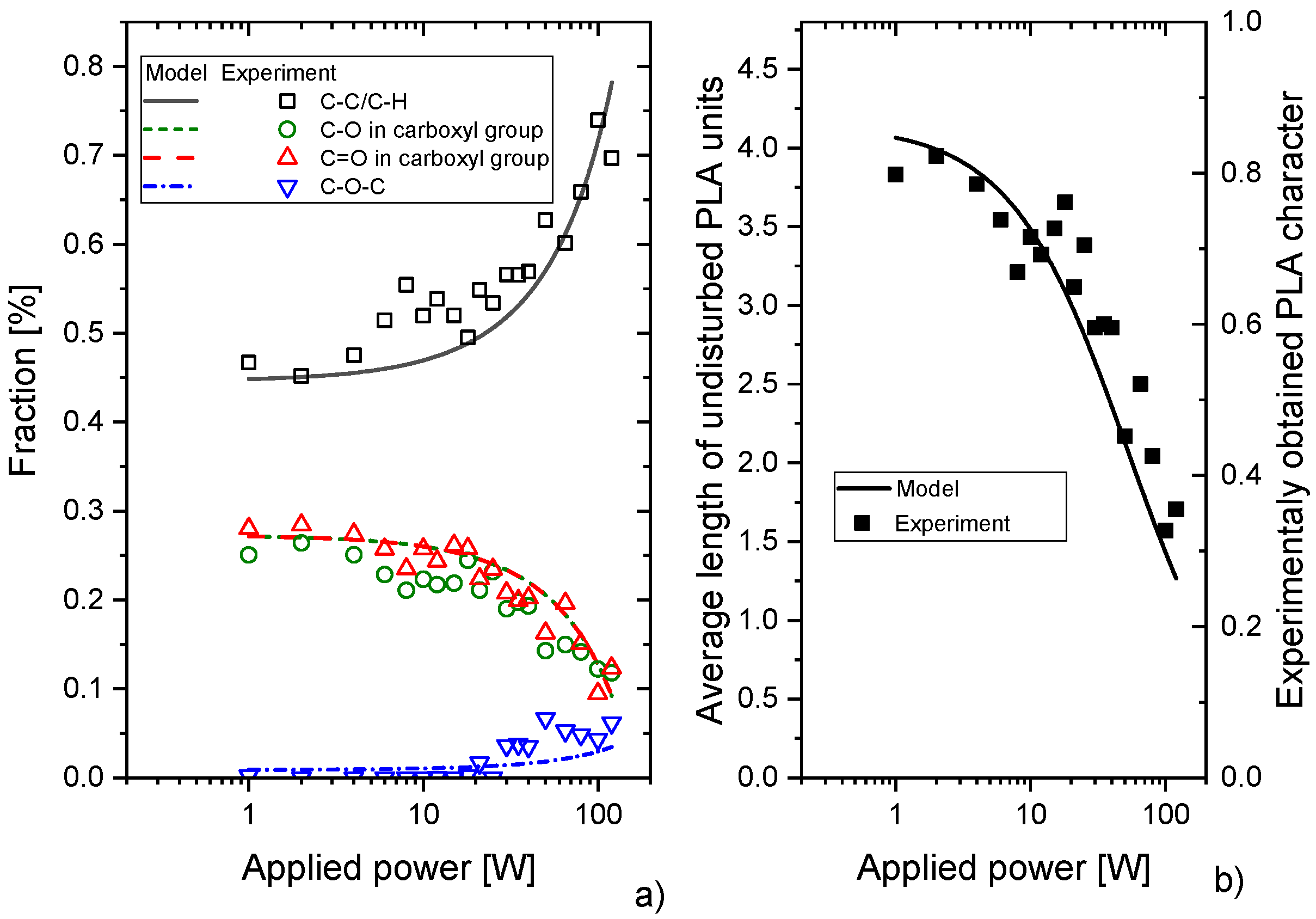
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, M.; Bell, A.T. A Review of Recent Advances in Plasma Polymerization. In Plasma Polymerization; American Chemical Society: Washington, DC, USA, 1979; Volume 108, pp. 1–33. ISBN 9780841205109. [Google Scholar]
- Friedrich, J. Mechanisms of plasma polymerization—Reviewed from a chemical point of view. Plasma Process. Polym. 2011, 8, 783–802. [Google Scholar] [CrossRef]
- Goodman, J. The formation of thin polymer films in the gas discharge. J. Polym. Sci. 1960, 44, 551–552. [Google Scholar] [CrossRef]
- Bewilogua, K.; Bräuer, G.; Dietz, A.; Gäbler, J.; Goch, G.; Karpuschewski, B.; Szyszka, B. Surface technology for automotive engineering. CIRP Ann. Manuf. Technol. 2009, 58, 608–627. [Google Scholar] [CrossRef]
- Wang, T.F.; Lin, T.J.; Yang, D.J.; Antonelli, J.A.; Yasuda, H.K. Corrosion protection of cold-rolled steel by low temperature plasma interface engineering I. Enhancement of E-coat adhesion. Prog. Org. Coat. 1996, 28, 291–297. [Google Scholar] [CrossRef]
- Grundmeier, G.; Thiemann, P.; Carpentier, J.; Barranco, V. Tailored thin plasma polymers for the corrosion protection of metals. Surf. Coat. Technol. 2003, 174, 996–1001. [Google Scholar] [CrossRef]
- Ogino, A.; Nagatsu, M. Gas barrier properties of hydrogenated amorphous carbon films coated on polymers by surface-wave plasma chemical vapor deposition. Thin Solid Films 2007, 515, 3597–3601. [Google Scholar] [CrossRef]
- Polonskyi, O.; Kylián, O.; Petr, M.; Choukourov, A.; Hanuš, J.; Biederman, H. Gas barrier properties of hydrogenated amorphous carbon films coated on polyethylene terephthalate by plasma polymerization in argon/n-hexane gas mixture. Thin Solid Films 2013, 540, 65–68. [Google Scholar] [CrossRef]
- Milella, A.; Di Mundo, R.; Palumbo, F.; Favia, P.; Fracassi, F.; d’Agostino, R. Plasma nanostructuring of polymers: Different routes to superhydrophobicity. Plasma Process. Polym. 2009, 6, 460–466. [Google Scholar] [CrossRef]
- Zille, A.; Oliveira, F.R.; Souto, P.A.P. Plasma treatment in textile industry. Plasma Process. Polym. 2015, 12, 98–131. [Google Scholar] [CrossRef] [Green Version]
- Cvelbar, U.; Walsh, J.L.; Černák, M.; de Vries, H.W.; Reuter, S.; Belmonte, T.; Corbella, C.; Miron, C.; Hojnik, N.; Jurov, A.; et al. White paper on the future of plasma science and technology in plastics and textiles. Plasma Process. Polym. 2019, 16, 1700228. [Google Scholar] [CrossRef] [Green Version]
- Tudoran, C.; Roşu, M.C.; Coroş, M. A concise overview on plasma treatment for application on textile and leather materials. Plasma Process. Polym. 2020, 17, 2000046. [Google Scholar] [CrossRef]
- Siow, K.S.; Britcher, L.; Kumar, S.; Griesser, H.J. Plasma methods for the generation of chemically reactive surfaces for biomolecule immobilization and cell colonization—A review. Plasma Process. Polym. 2006, 3, 392–418. [Google Scholar] [CrossRef]
- Bhatt, S.; Pulpytel, J.; Mirshahi, M.; Arefi-Khonsari, F. Plasma co-polymerized nano coatings—As a biodegradable solid carrier for tunable drug delivery applications. Polymer 2013, 54, 4820–4829. [Google Scholar] [CrossRef]
- Kratochvíl, J.; Kahoun, D.; Štěrba, J.; Langhansová, H.; Lieskovská, J.; Fojtíková, P.; Hanuš, J.; Kousal, J.; Kylián, O.; Straňák, V. Plasma polymerized C:H:N:O thin films for controlled release of antibiotic substances. Plasma Process. Polym. 2018, 15, 1700160. [Google Scholar] [CrossRef]
- Bekeschus, S.; Favia, P.; Robert, E.; von Woedtke, T. White paper on plasma for medicine and hygiene: Future in plasma health sciences. Plasma Process. Polym. 2019, 16, 1800033. [Google Scholar] [CrossRef] [Green Version]
- Hegemann, D.; Nisol, B.; Watson, S.; Wertheimer, M.R. Energy Conversion Efficiency in Plasma Polymerization—A Comparison of Low- and Atmospheric-Pressure Processes. Plasma Process. Polym. 2016, 13, 834–842. [Google Scholar] [CrossRef]
- Choukourov, A.; Hanuš, J.; Kousal, J.; Grinevich, A.; Pihosh, Y.; Slavínská, D.; Biederman, H. Thin polymer films from polyimide vacuum thermal degradation with and without a glow discharge. Vacuum 2006, 80, 923–929. [Google Scholar] [CrossRef]
- Choukourov, A.; Gordeev, I.; Polonskyi, O.; Artemenko, A.; Hanyková, L.; Krakovský, I.; Kylián, O.; Slavínská, D.; Biederman, H. Polyethylene (ethylene oxide)-like plasma polymers produced by plasma-assisted vacuum evaporation. Plasma Process. Polym. 2010, 7, 445–458. [Google Scholar] [CrossRef]
- Choukourov, A.; Grinevich, A.; Polonskyi, O.; Hanus, J.; Kousal, J.; Slavinska, D.; Biederman, H. Vacuum thermal degradation of poly(ethylene oxide). J. Phys. Chem. B 2009, 113, 2984–2989. [Google Scholar] [CrossRef] [PubMed]
- Choukourov, A.; Gordeev, I.; Arzhakov, D.; Artemenko, A.; Kousal, J.; Kylián, O.; Slavínská, D.; Biederman, H. Does cross-link density of PEO-like plasma polymers influence their resistance to adsorption of fibrinogen? Plasma Process. Polym. 2012, 9, 48–58. [Google Scholar] [CrossRef]
- Choukourov, A.; Hanuš, J.; Kousal, J.; Grinevich, A.; Pihosh, Y.; Slavínská, D.; Biederman, H. Plasma polymer films from sputtered polyimide. Vacuum 2006, 81, 517–526. [Google Scholar] [CrossRef]
- Kousal, J.; Sedlaříková, J.; Kolářová Rašková, Z.; Krtouš, Z.; Kučerová, L.; Hurajová, A.; Vaidulych, M.; Hanuš, J.; Lehocký, M. Degradable poly(ethylene oxide)-like plasma polymer films used for the controlled release of nisin. Polymers 2020, 12, 1263. [Google Scholar] [CrossRef] [PubMed]
- Kousal, J.; Kolářová Rašková, Z.; Sedlaříková, J.; Stloukal, P.; Solař, P.; Krtouš, Z.; Choukourov, A.; Sedlařík, V.; Lehocký, M. Plasma polymers with controlled degradation behaviour. In Proceedings of the NANOCON 2017—Conference Proceedings, 9th International Conference on Nanomaterials—Research and Application, Brno, Czech Republic, 18–20 October 2017; TANGER Ltd.: Ostrava, Czech Republic, 2018; pp. 461–466. [Google Scholar]
- Kousal, J.; Krtouš, Z.; Kolářová Rašková, Z.; Sedlaříková, J.; Schäfer, J.; Kučerová, L.; Shelemin, A.; Solař, P.; Hurajová, A.; Biederman, H.; et al. Degradable plasma polymer films with tailored hydrolysis behavior. Vacuum 2020, 173, 109062. [Google Scholar] [CrossRef]
- Vink, E.T.H.; Rábago, K.R.; Glassner, D.A.; Springs, B.; O’Connor, R.P.; Kolstad, J.; Gruber, P.R. The sustainability of nature worksTM polylactide polymers and ingeoTM polylactide fibers: An update of the future. Initiated by the 1st International Conference on Bio-based Polymers (ICBP 2003), 12–14 November 2003, Saitama, Japan. Macromol. Biosci. 2004, 4, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Morent, R.; De Geyter, N.; Desmet, T.; Dubruel, P.; Leys, C. Plasma surface modification of biodegradable polymers: A review. Plasma Process. Polym. 2011, 8, 171–190. [Google Scholar] [CrossRef]
- Pawar, R.P.; Tekale, S.U.; Shisodia, S.U.; Totre, J.T.; Domb, A.J. Biomedical applications of poly(lactic acid). Rec. Pat. Regen. Med. 2014, 4, 40–51. [Google Scholar] [CrossRef]
- DeStefano, V.; Khan, S.; Tabada, A. Applications of PLA in modern medicine. Eng. Regen. 2020, 1, 76–87. [Google Scholar] [CrossRef]
- Casalini, T.; Rossi, F.; Castrovinci, A.; Perale, G. A Perspective on Polylactic Acid-Based Polymers Use for Nanoparticles Synthesis and Applications. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef]
- Ligot, S.; Renaux, F.; Denis, L.; Cossement, D.; Nuns, N.; Dubois, P.; Snyders, R. Experimental study of the plasma polymerization of ethyl lactate. Plasma Process. Polym. 2013, 10, 999–1009. [Google Scholar] [CrossRef]
- Ligot, S.; Bousser, E.; Cossement, D.; Klemberg-Sapieha, J.; Viville, P.; Dubois, P.; Snyders, R. Correlation between mechanical properties and cross-linking degree of ethyl lactate plasma polymer films. Plasma Process. Polym. 2015, 12, 508–518. [Google Scholar] [CrossRef]
- Laurent, M.; Desjardins, E.; Meichelboeck, M.; Naudé, N.; Stafford, L.; Gherardi, N.; Laroche, G. Characterization of argon dielectric barrier discharges applied to ethyl lactate plasma polymerization. J. Phys. D Appl. Phys. 2017, 50, 475205. [Google Scholar] [CrossRef]
- Nisol, B.; Watson, S.; Lerouge, S.; Wertheimer, M.R. Energetics of reactions in a dielectric barrier discharge with argon carrier gas: IV ethyl lactate. Plasma Process. Polym. 2016, 13, 965–969. [Google Scholar] [CrossRef]
- Kucharczyk, P.; Hnatkova, E.; Dvorak, Z.; Sedlarik, V. Novel aspects of the degradation process of PLA based bulky samples under conditions of high partial pressure of water vapour. Polym. Degrad. Stab. 2013, 98, 150–157. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1991, 93, 151–170. [Google Scholar] [CrossRef]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA 300 Database; John Wiley & Sons: Chichester, UK, 1992; ISBN 047193592. [Google Scholar]
- Kiss, É.; Bertóti, I.; Vargha-Butler, E.I. XPS and wettability characterization of modified poly(lactic acid) and poly(lactic/glycolic acid) films. J. Colloid Interface Sci. 2002, 245, 91–98. [Google Scholar] [CrossRef]
- Kousal, J.; Hanuš, J.; Choukourov, A.; Polonskyi, O.; Biederman, H.; Slavínská, D. In situ diagnostics of RF magnetron sputtering of nylon. Plasma Process. Polym. 2009, 6, 803–812. [Google Scholar] [CrossRef]
- Zou, H.; Yi, C.; Wang, L.; Liu, H.; Xu, W. Thermal degradation of poly(lactic acid) measured by thermogravimetry coupled to Fourier transform infrared spectroscopy. J. Therm. Anal. Calorim. 2009, 97, 929–935. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krtouš, Z.; Hanyková, L.; Krakovský, I.; Nikitin, D.; Pleskunov, P.; Kylián, O.; Sedlaříková, J.; Kousal, J. Structure of Plasma (re)Polymerized Polylactic Acid Films Fabricated by Plasma-Assisted Vapour Thermal Deposition. Materials 2021, 14, 459. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14020459
Krtouš Z, Hanyková L, Krakovský I, Nikitin D, Pleskunov P, Kylián O, Sedlaříková J, Kousal J. Structure of Plasma (re)Polymerized Polylactic Acid Films Fabricated by Plasma-Assisted Vapour Thermal Deposition. Materials. 2021; 14(2):459. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14020459
Chicago/Turabian StyleKrtouš, Zdeněk, Lenka Hanyková, Ivan Krakovský, Daniil Nikitin, Pavel Pleskunov, Ondřej Kylián, Jana Sedlaříková, and Jaroslav Kousal. 2021. "Structure of Plasma (re)Polymerized Polylactic Acid Films Fabricated by Plasma-Assisted Vapour Thermal Deposition" Materials 14, no. 2: 459. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14020459