
Multidimensional Ln-Aminophthalate Photoluminescent Coordination Polymers

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
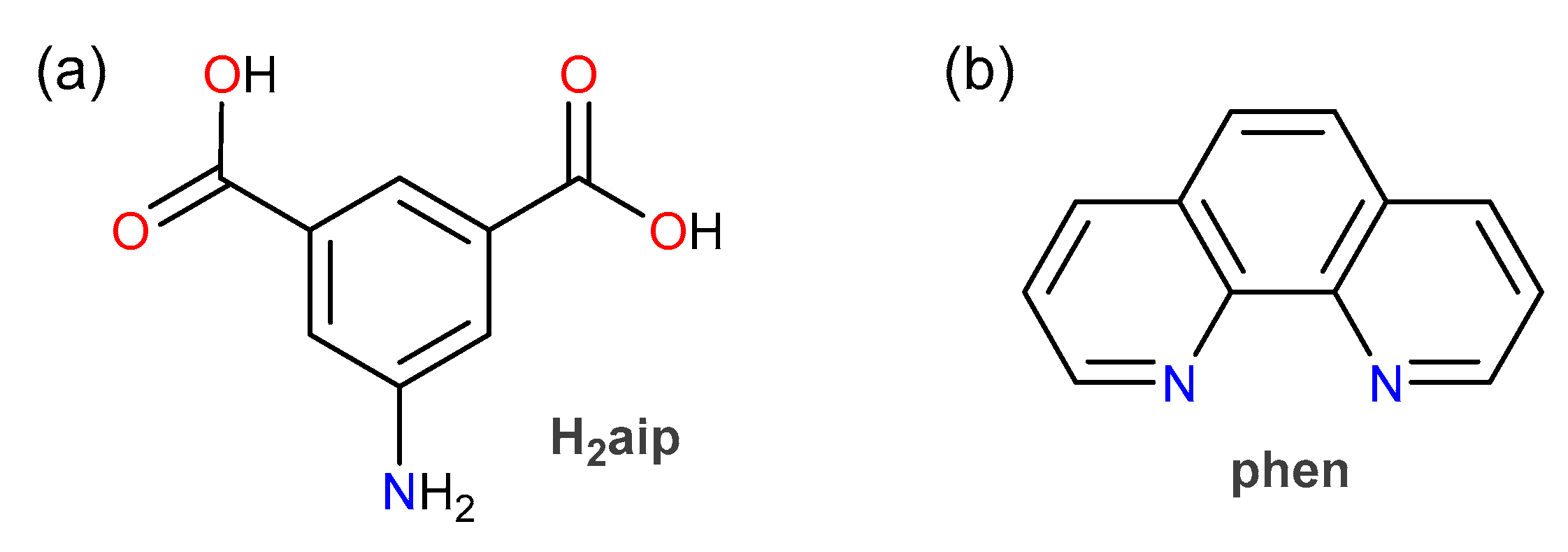
2.1. Materials
2.2. Coordination Polymers’ Preparation
2.3. Single Crystal X-Ray Diffraction
2.4. Materials Characterization
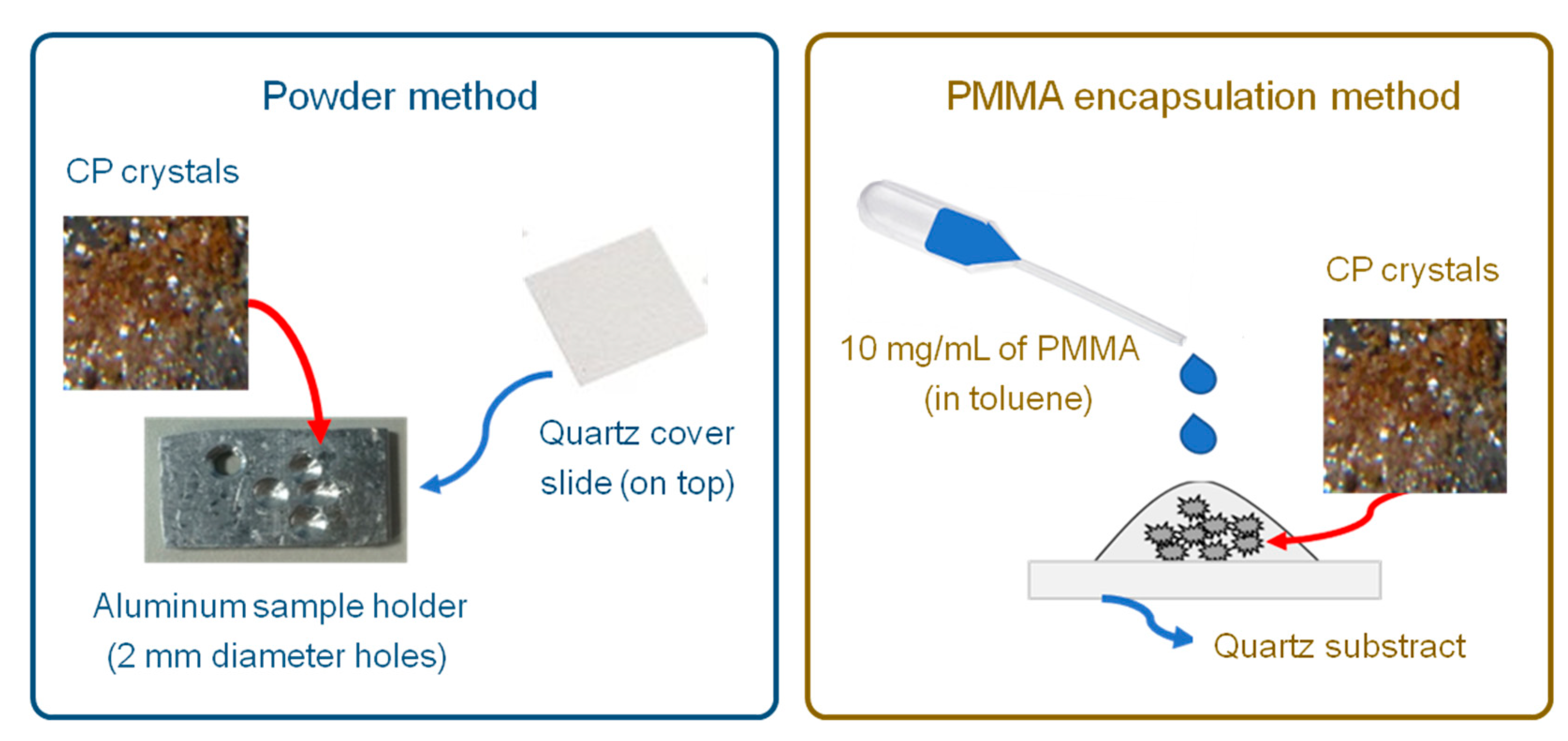
2.5. Photoluminescence Characterization
3. Results and Discussion
3.1. Coordination Polymers Preparation
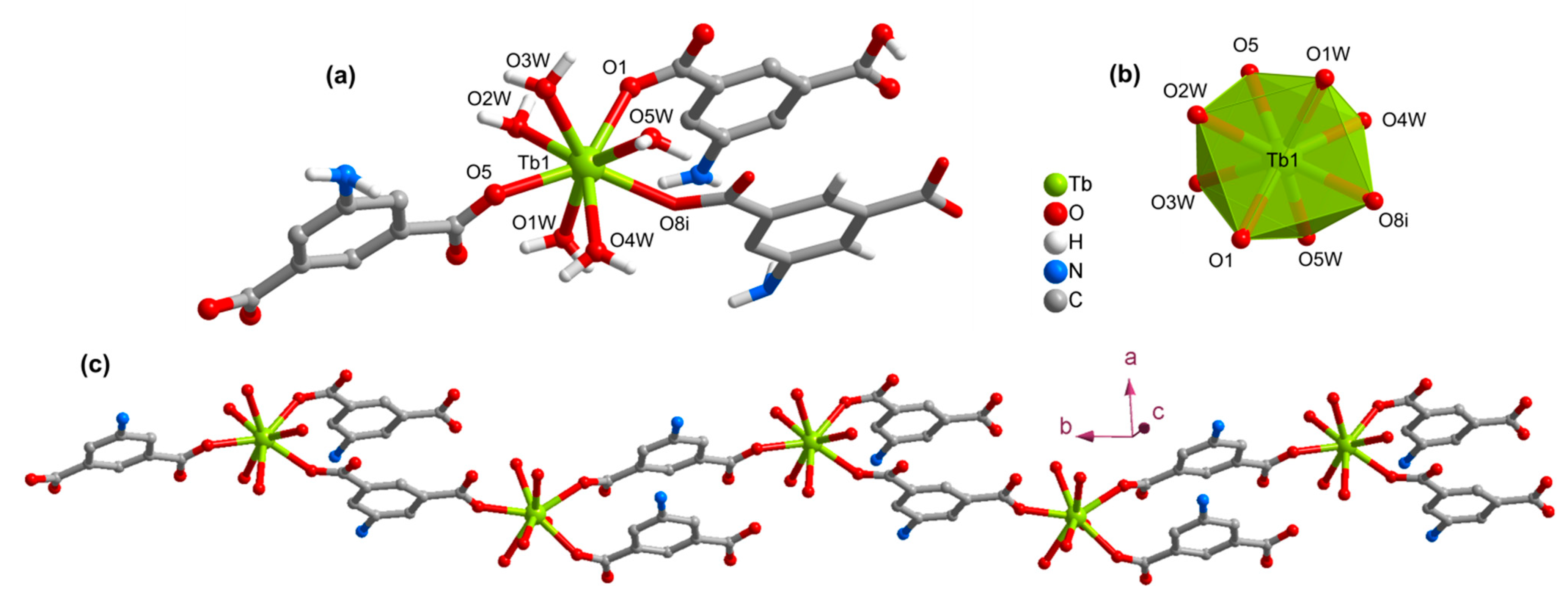
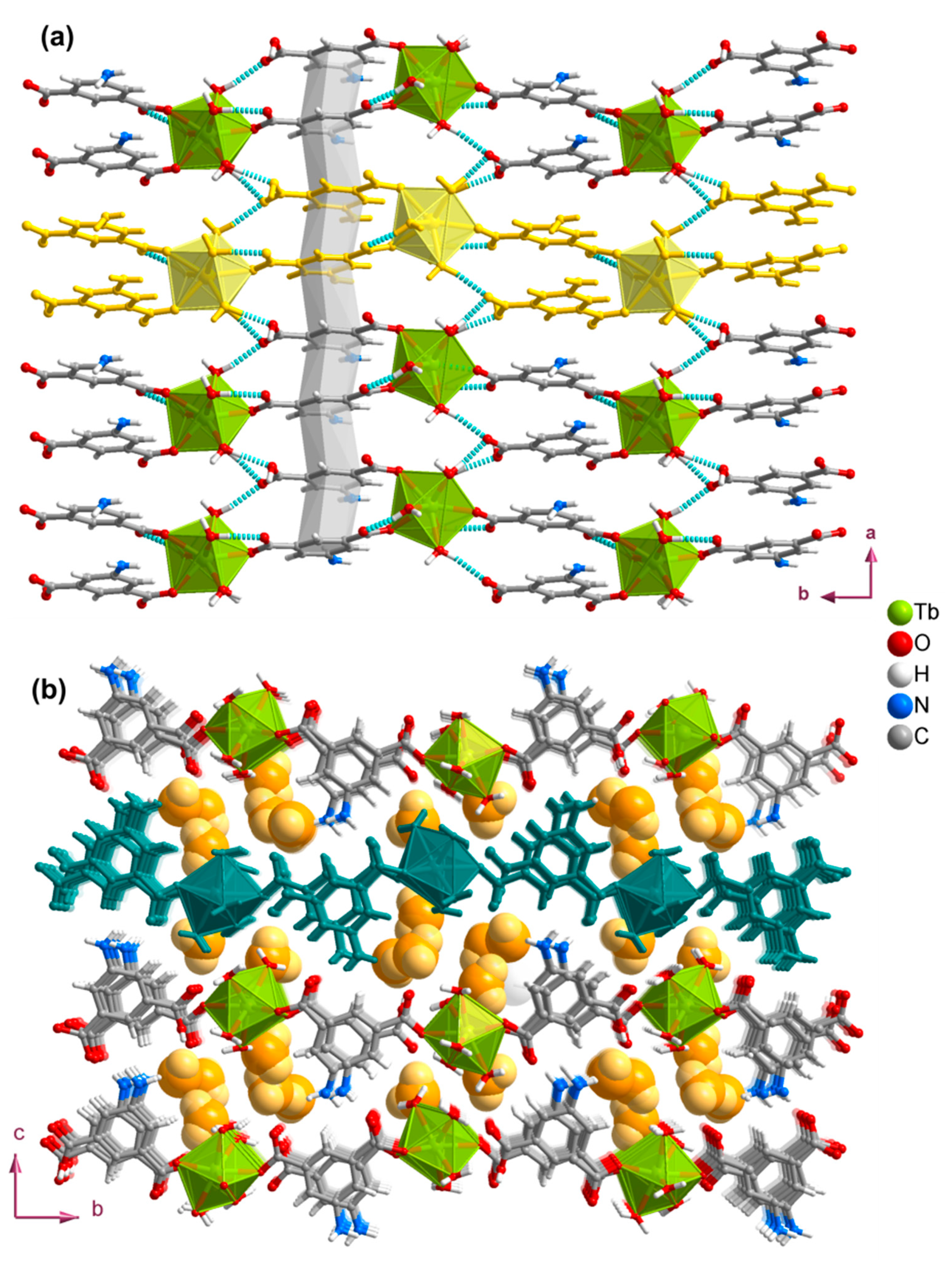
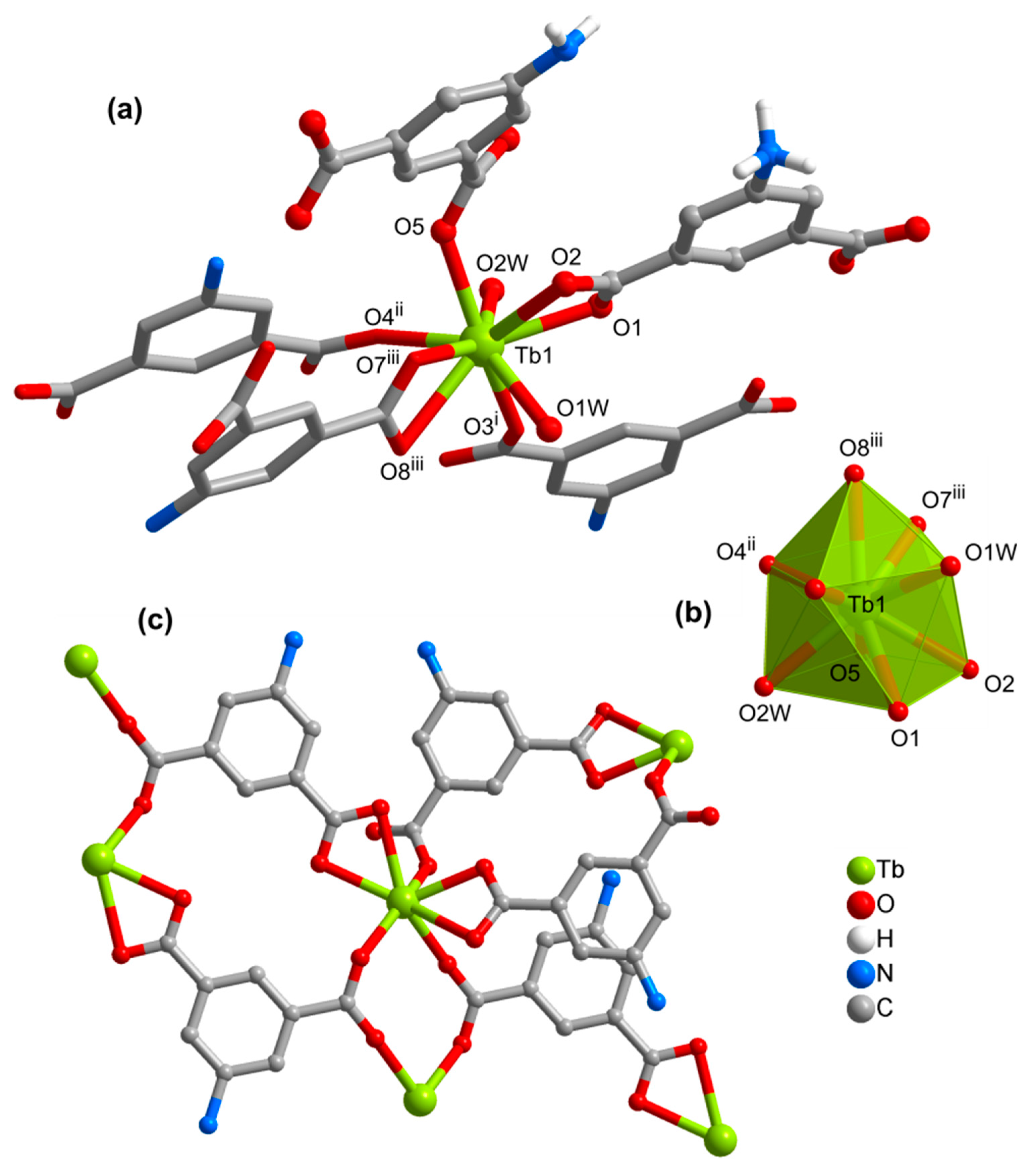
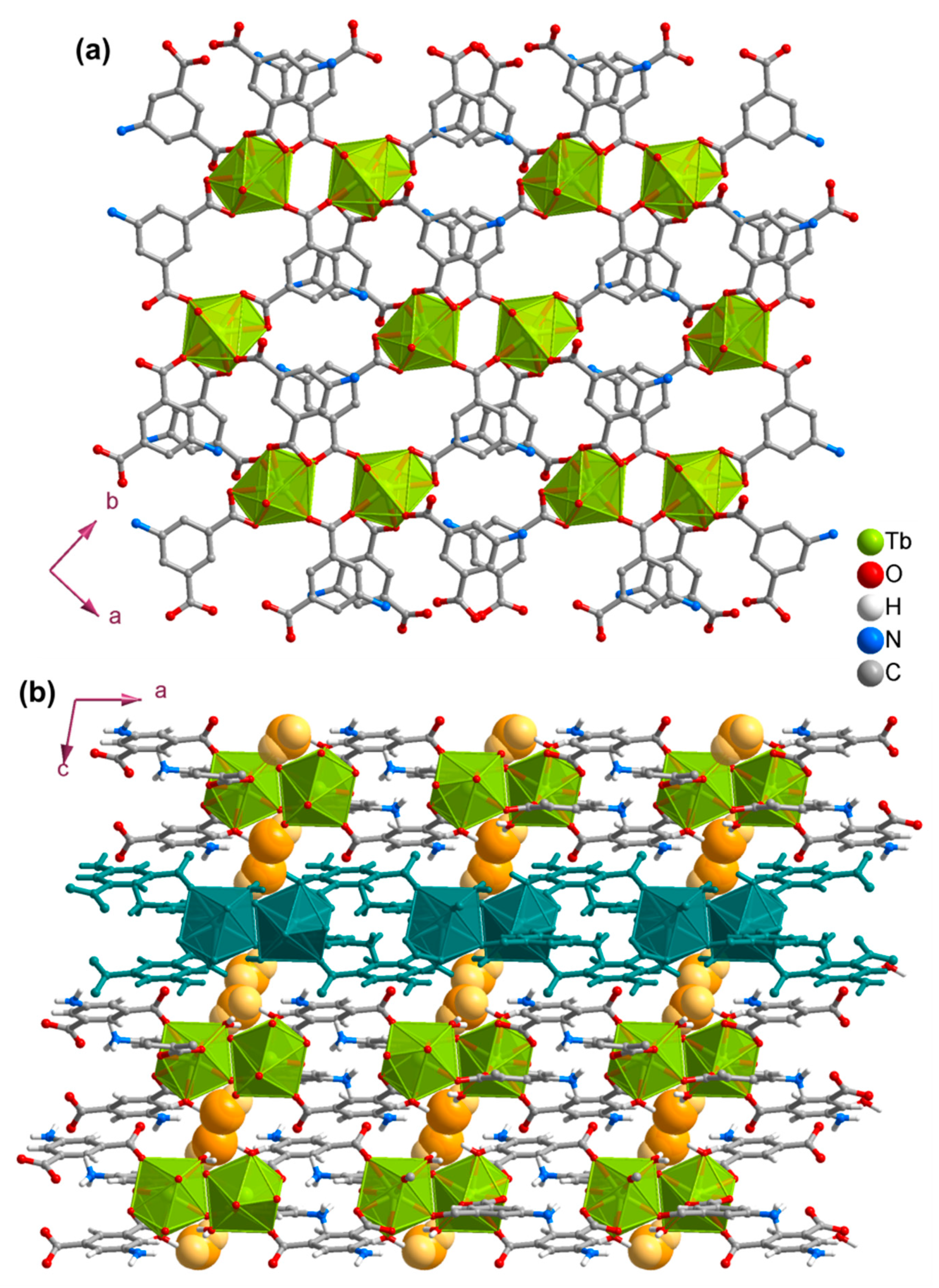
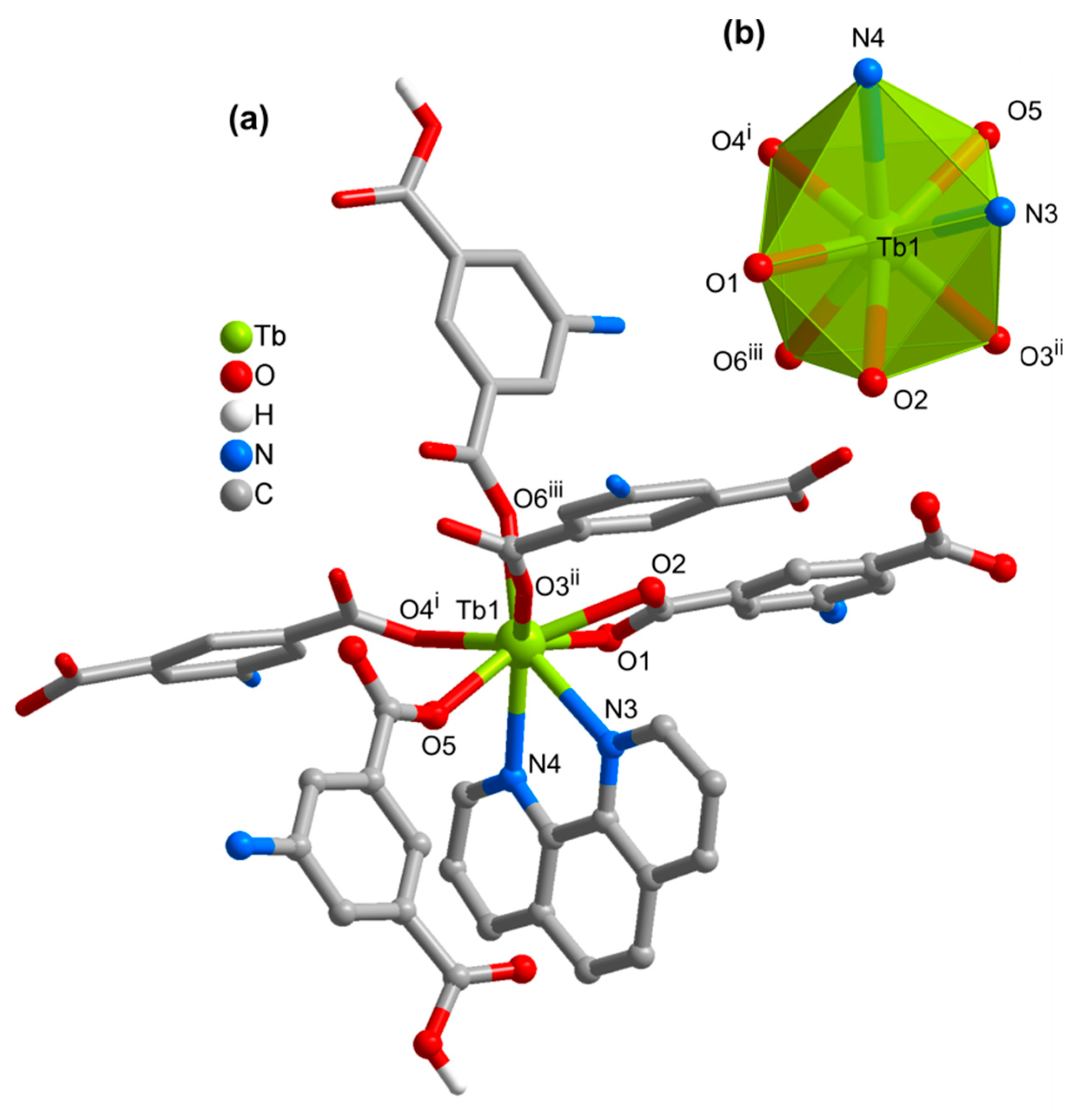
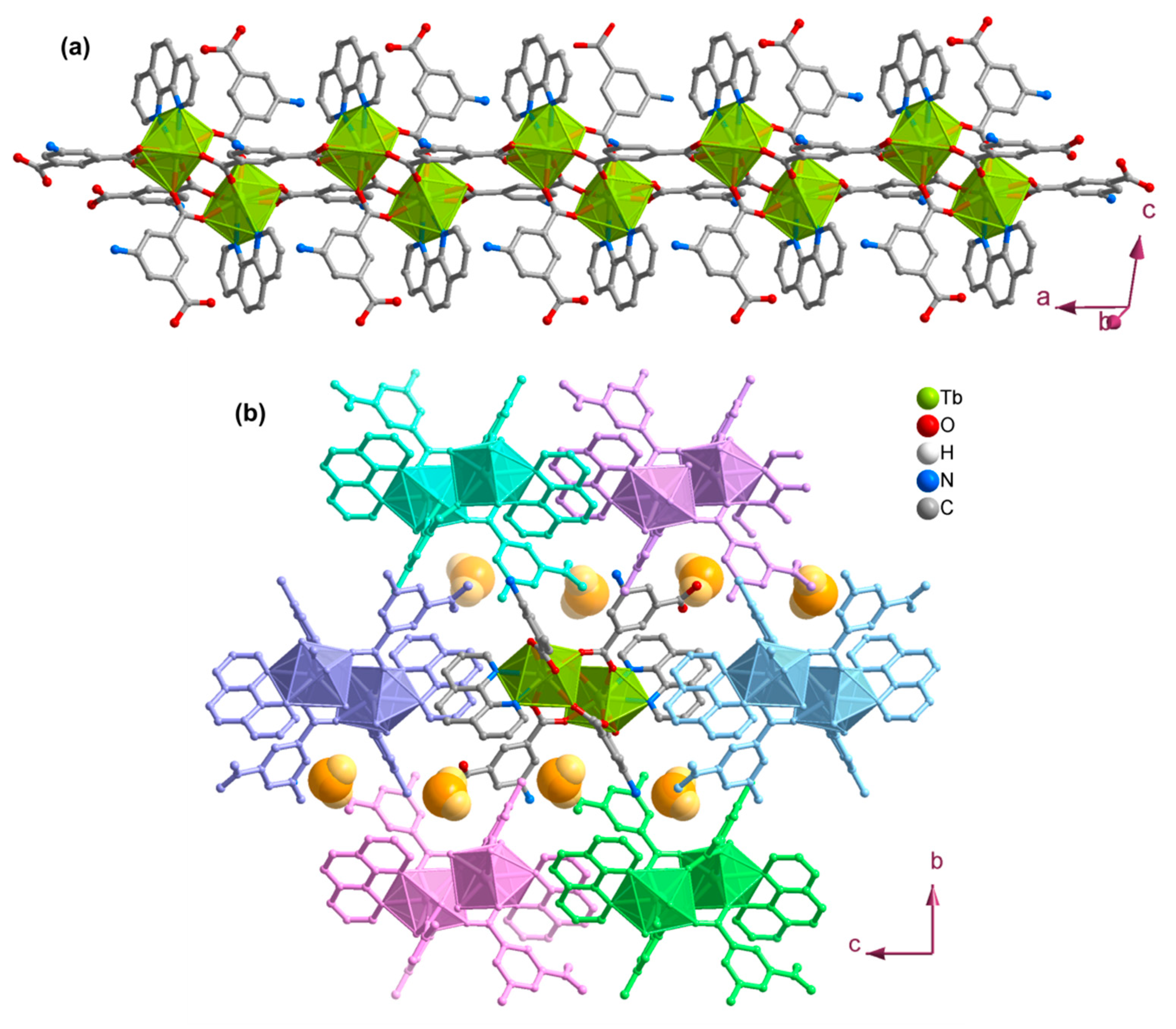
3.2. Structures of the Coordination Polymers
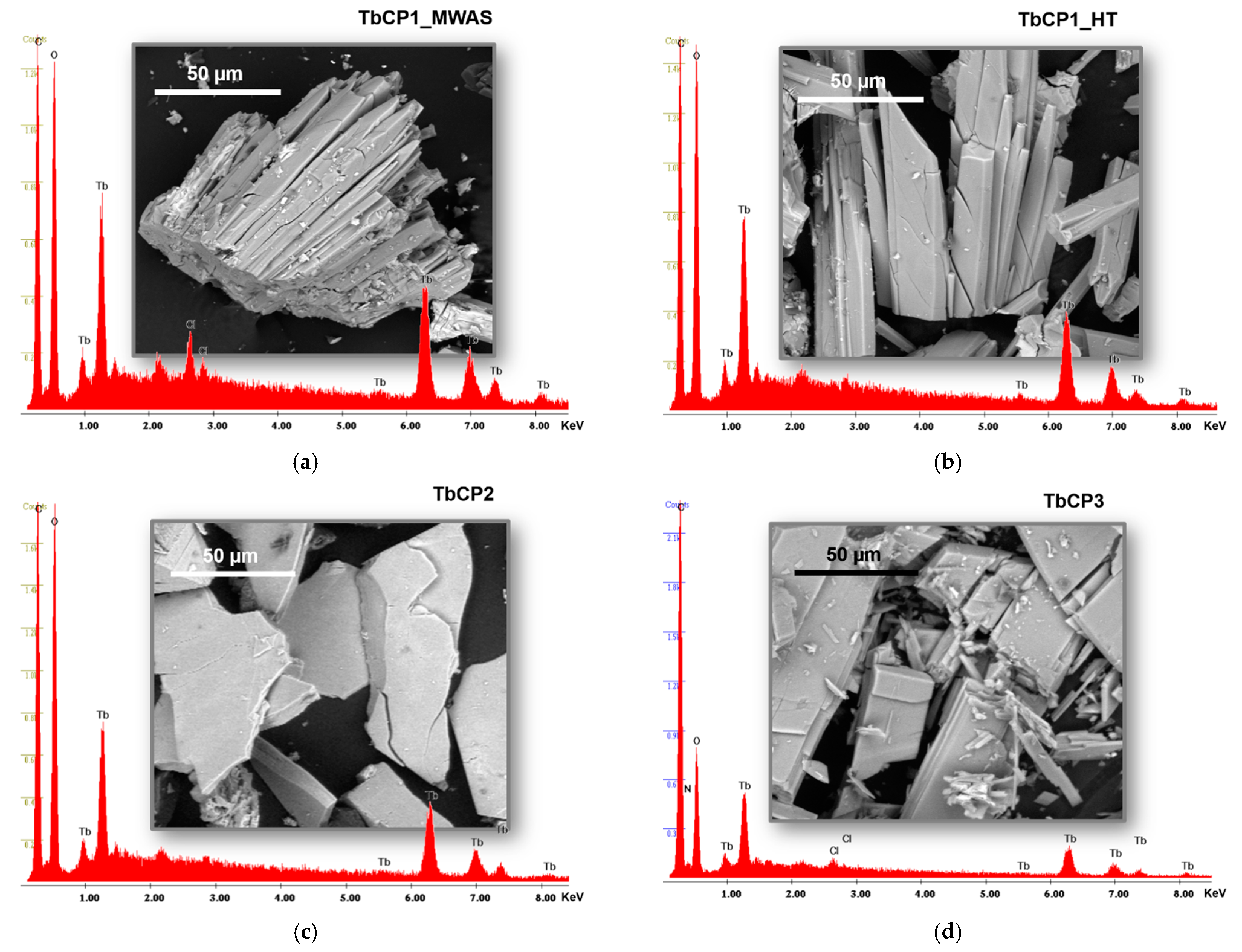
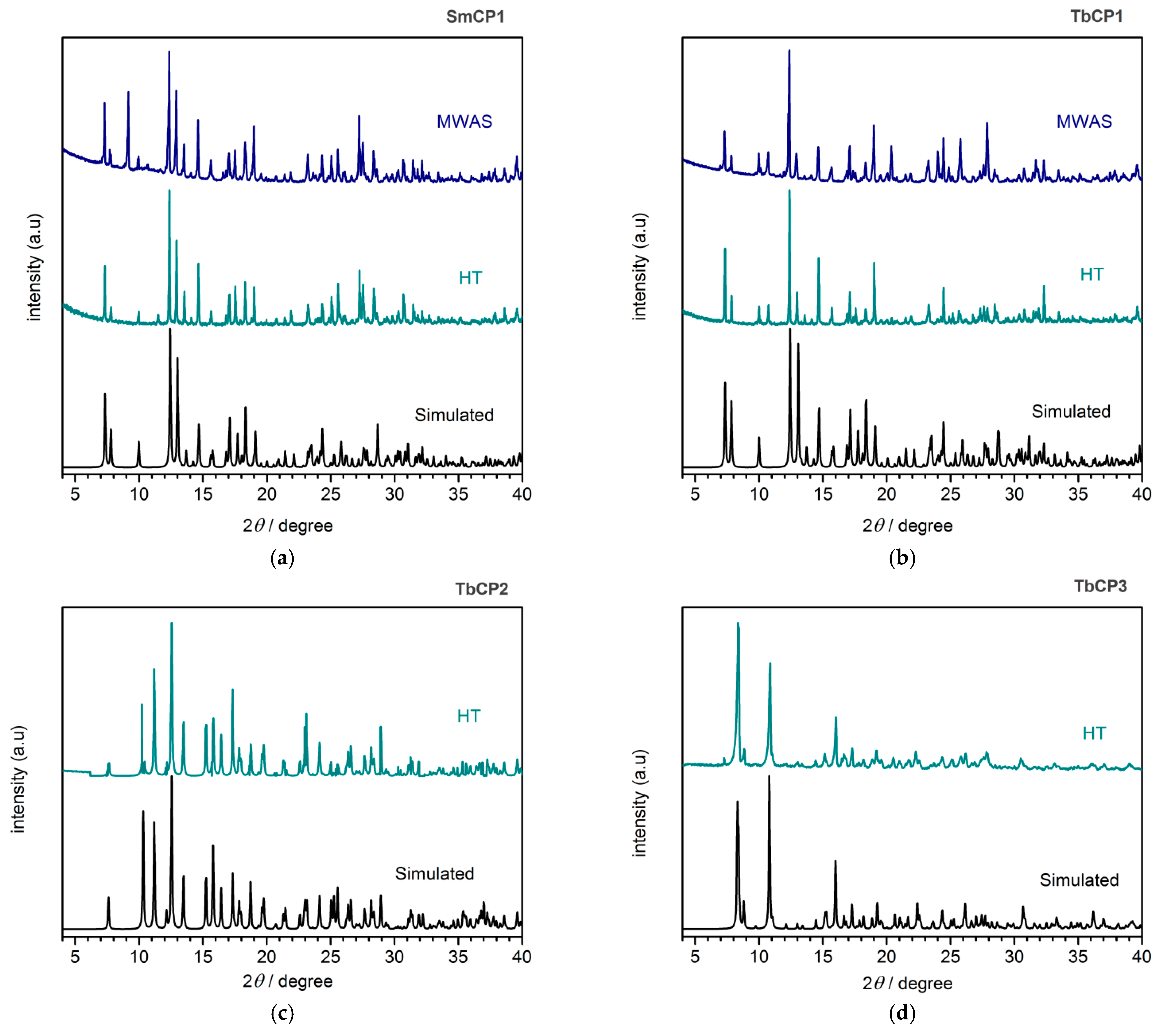
3.3. Crystalline and Morphology Analysis
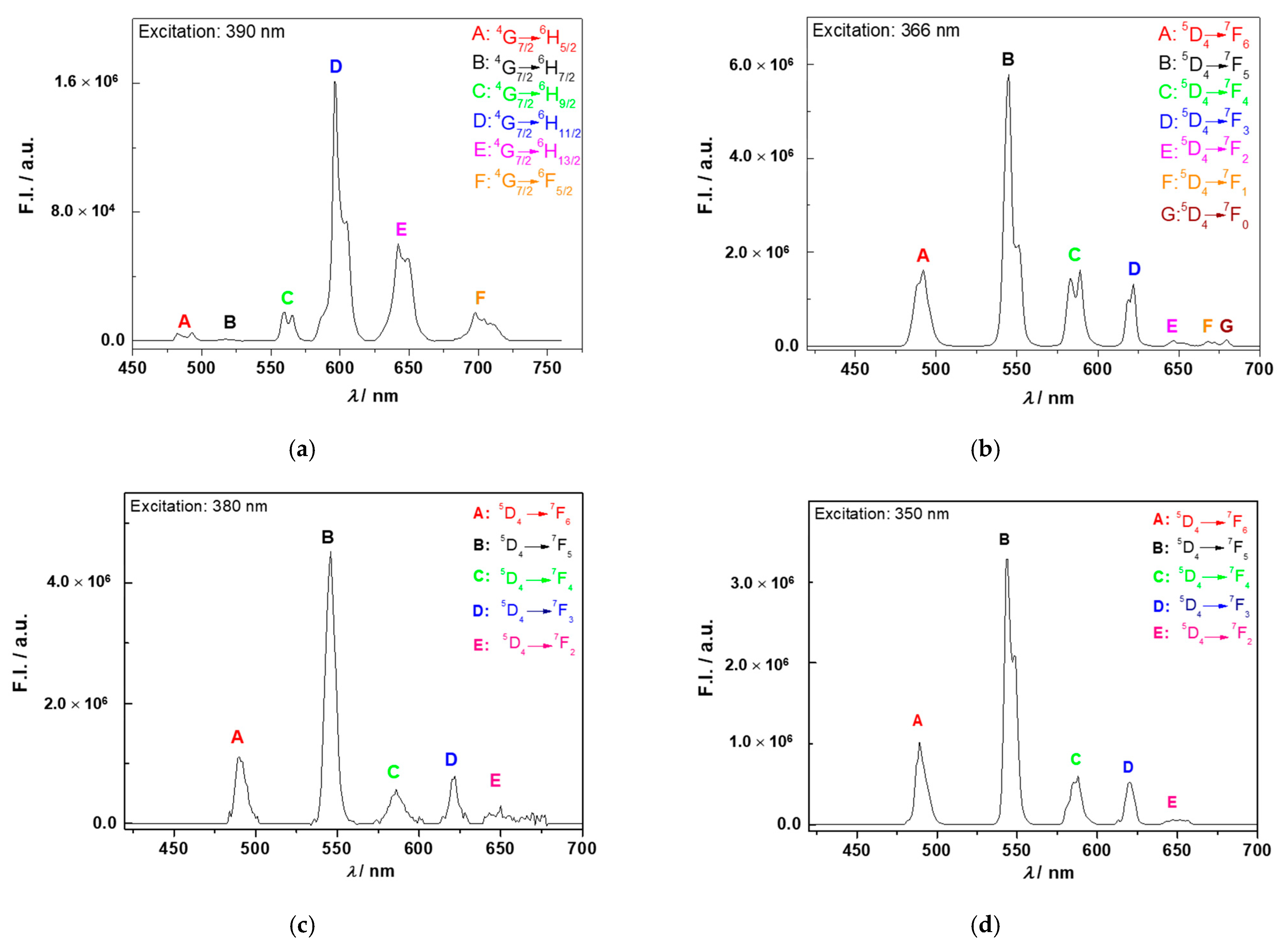
3.4. Photoluminescence Studies
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bunzli, J.C.G.; Eliseeva, S.V. Intriguing aspects of lanthanide luminescence. Chem. Sci. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Utochnikova, V.V.; Kuzmina, N.P. Photoluminescence of lanthanide aromatic carboxylates. Russ. J. Coord. Chem. 2016, 42, 679–694. [Google Scholar] [CrossRef]
- Heine, J.; Müller-Buschbaum, K. Engineering metal-based luminescence in coordination polymers and metal–organic frameworks. Chem. Soc. Rev. 2013, 42, 9232–9242. [Google Scholar] [CrossRef]
- Einkauf, J.D.; Clark, J.M.; Paulive, A.; Tanner, G.P.; de Lill, D.T. A general model of sensitized luminescence in lanthanide-based coordination polymers and metal–organic framework materials. Inorg. Chem. 2017, 56, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Gorai, T.; Schmitt, W.; Gunnlaugsson, T. Highlights of the development and application of luminescent lanthanide based coordination polymers, MOFs and functional nanomaterials. Dalton Trans. 2021, 50, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, X.; Jiang, Y.; Hou, Z.; Cheng, Z.; Ma, P.; Li, C.; Lin, J. Highly luminescent lanthanide fluoride nanoparticles functionalized by aromatic carboxylate acids. RSC Adv. 2014, 4, 55100–55107. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, S.; Qu, X.; Yang, Q.; Liu, X.; Wei, Q.; Xie, G.; Chen, S. Synthesis, crystal structure and photoluminescence property of Eu/Tb MOFs with mixed polycarboxylate ligands. J. Solid State Chem. 2015, 231, 223–229. [Google Scholar] [CrossRef]
- Zhou, W.; Wu, Y.-P.; Zhou, Z.-H.; Qin, Z.-S.; Ye, X.; Tian, F.-Y.; Li, D.-S. Construction of a series of lanthanide metal–organic frameworks (Ln-MOFs) based on a new symmetrical penta-aromatic carboxylate strut: Structure, luminescent and magnetic properties. Inorg. Chim. Acta 2016, 453, 757–763. [Google Scholar] [CrossRef]
- Chen, Q.; Cheng, J.; Wang, J.; Li, L.; Liu, Z.; Zhou, X.; You, Y.; Huang, W. A fluorescent Eu(III) MOF for highly selective and sensitive sensing of picric acid. Sci. China Chem. 2019, 62, 205–211. [Google Scholar] [CrossRef]
- Yang, G.L.; Jiang, X.L.; Xu, H.; Zhao, B. Applications of MOFs as luminescent sensors for environmental pollutants. Small 2021, 202005327. [Google Scholar] [CrossRef]
- Jin, J.; Xue, J.J.; Liu, Y.C.; Yang, G.P.; Wang, Y.Y. Recent progresses in luminescent metal-organic frameworks (LMOFs) as sensors for the detection of anions and cations in aqueous solution. Dalton Trans. 2021, 50, 1950–1972. [Google Scholar] [CrossRef]
- D’Vries, R.F.; Gomez, G.E.; Hodak, J.H.; Soler-Illia, G.J.A.A.; Ellena, J. Tuning the structure, dimensionality and luminescent properties of lanthanide metal-organic frameworks under ancillary ligand influence. Dalton Trans. 2016, 45, 646–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.L.; Qu, X.L.; Li, Z.; Li, X.; Sun, H.L. Isostructural lanthanide-based metal-organic frameworks: Structure, photoluminescence and magnetic properties. Dalton Trans. 2017, 47, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Liu, Z.Q.; Huang, Y.; Chen, F.; Zeng, C.H.; Zhong, S.; Ng, S.W. Highly luminescent Ln-MOFs based on 1,3-adamantanediacetic acid as bifunctional sensor. Sens. Actuators B Chem. 2018, 257, 705–713. [Google Scholar] [CrossRef]
- Xu, Q.-W.; Dong, G.; Cui, R.; Li, X. 3D lanthanide-coordination frameworks constructed by a ternary mixed-ligand: Crystal structure, luminescence and luminescence sensing. CrystEngComm 2020, 22, 740–750. [Google Scholar] [CrossRef]
- Liu, J.; Hang, M.; Wu, D.; Jin, J.; Cheng, J.-G.; Yang, G.; Wang, Y.-Y. Fine-tuning the porosities of the entangled isostructural Zn(II)-based metal–organic frameworks with active sites by introducing different N-auxiliary ligands: Selective gas sorption and efficient CO2 conversion. Inorg. Chem. 2020, 59, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Kariem, M.; Kumar, M.; Yawer, M.; Sheikh, H.N. Solvothermal synthesis and structure of coordination polymers of Nd(III) and Dy(III) with rigid isophthalic acid derivatives and flexible adipic acid. J. Mol. Struct. 2017, 1150, 438–446. [Google Scholar] [CrossRef]
- Chandran, P.S.; Mol, U.S.; Drisya, R.; Sudarsanakumar, M.R.; Kurup, M.P. Structural studies of poly[(μ2-acetato)(μ3-5-aminoisophthalato)diaquacerium(III) monohydrate]: A new three dimensional fluorescent metal-organic framework constructed from dimers of CeO9 polyhedra with hydrophilic ‘S’ shaped channels. J. Mol. Struct. 2017, 1137, 396–402. [Google Scholar] [CrossRef]
- Winkless, L.; Tan, R.H.C.; Zheng, Y.; Motevalli, M.; Wyatt, P.B.; Gillin, W.P. Quenching of Er(III) luminescence by ligand C–H vibrations: Implications for the use of erbium complexes in telecommunications. Appl. Phys. Lett. 2006, 89, 111115. [Google Scholar] [CrossRef] [Green Version]
- Bischof, C.; Wahsner, J.; Scholten, J.; Trosien, S.; Seitz, M. Quantification of C−H quenching in near-IR luminescent Ytterbium and Neodymium cryptates. J. Am. Chem. Soc. 2010, 132, 14334–14335. [Google Scholar] [CrossRef] [PubMed]
- Einkauf, J.D.; Kelley, T.T.; Chan, B.C.; de Lill, D.T. Rethinking sensitized luminescence in lanthanide coordination polymers and MOFs: Band sensitization and water enhanced Eu luminescence in [Ln(C15H9O5)3(H2O)3]n (Ln = Eu, Tb). Inorg. Chem. 2016, 55, 7920–7927. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Arata, R.; Ogawa, T.; Yoshida, M.; Kato, M. Effect of water coordination on luminescent properties of pyrazine-bridged dinuclear Cu(I) complexes. Inorg. Chem. 2017, 56, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, Y.; Ma, Y.; Li, L. Dual-functional recyclable luminescent sensors based on 2D lanthanide-based metal-organic frameworks for highly sensitive detection of Fe3+ and 2,4-dinitrophenol. Dye. Pigment. 2017, 146, 263–271. [Google Scholar] [CrossRef]
- Du, Y.; Yang, H.; Liu, R.; Shao, C.; Yang, L. A multi-responsive chemosensor for highly sensitive and selective detection of Fe3+, Cu2+, Cr2O72-and nitrobenzene based on a luminescent lanthanide metal-organic framework. Dalton Trans. 2020, 49, 13003–13016. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hou, S.L.; Zhao, B. Bimetallic lanthanide-organic framework membranes as a self-calibrating luminescent sensor for rapidly detecting antibiotics in water. ACS Appl. Mater. Interfaces 2020, 12, 38124–38131. [Google Scholar] [CrossRef]
- Huang, H.; Gao, W.; Zhang, X.M.; Zhou, A.M.; Liu, J.P. 3D LnIII-MOFs: Displaying slow magnetic relaxation and highly sensitive luminescence sensing of alkylamines. CrystEngComm 2019, 21, 694–702. [Google Scholar] [CrossRef]
- Ma, A.; Ke, F.; Jiang, J.; Yuan, Q.; Luo, Z.; Liu, J.; Kumar, A. Two lanthanide-based metal-organic frameworks for highly efficient adsorption and removal of fluoride ions from water. CrystEngComm 2017, 19, 2172–2177. [Google Scholar] [CrossRef]
- Yan, B.; Bai, Y.; Chen, Z. Synthesis, structure and luminescence of novel 1D chain coordination polymers [Ln(isophth)(Hisophth)(H2O)4·4H2O]n (Ln = Sm, Dy). J. Mol. Struct. 2005, 741, 141–147. [Google Scholar] [CrossRef]
- Sarma, D.; Prabu, M.; Biju, S.; Reddy, M.L.P.; Natarajan, S. Synthesis, structure and optical studies of a family of three-dimensional rare-earth aminoisophthalates [M(μ2-OH)(C8H5NO4)] (M = Y3+, La3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Dy3+ and Er3+). Eur. J. Inorg. Chem. 2010, 2010, 3813–3822. [Google Scholar] [CrossRef]
- Ma, M.-L.; Ji, C.; Zang, S.-Q. Syntheses, structures, tunable emission and white light emitting Eu3+ and Tb3+ doped lanthanide metal–organic framework materials. Dalton Trans. 2013, 42, 10579–10586. [Google Scholar] [CrossRef]
- Song, T.; Rao, X.; Cui, Y.; Yang, Y.; Qian, G. Synthesis and luminescent properties of color-tunable lanthanide complexes with 5-(pyridin-4-yl)isophthalic acid. J. Alloys Compd. 2013, 555, 22–27. [Google Scholar] [CrossRef]
- Liu, J.; Sun, W.; Liu, Z. White-light emitting materials with tunable luminescence based on steady Eu(III) doping of Tb(III) metal-organic frameworks. RSC Adv. 2016, 6, 25689–25694. [Google Scholar] [CrossRef]
- Kyprianidou, E.J.; Lazarides, T.; Kaziannis, S.; Kosmidis, C.; Itskos, G.; Manos, M.J.; Tasiopoulos, A.J. Single crystal coordinating solvent exchange as a general method for the enhancement of the photoluminescence properties of lanthanide MOFs. J. Mater. Chem. A 2014, 2, 5258–5266. [Google Scholar] [CrossRef]
- Zhao, S.; Xiao, J.; Zheng, T.; Liu, M.; Wu, H.; Liu, Z. Highly selective and sensitive detection of PO43- ions in aqueous solution by a luminescent terbium metal-organic framework. ACS Omega 2019, 4, 16378–16384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscoso, F.G.; Almeida, J.; Sousaraei, A.; Lopes-Costa, T.; Silva, A.M.G.; Cabanillas-Gonzalez, J.; Cunha-Silva, L.; Pedrosa, J.M. A lanthanide MOF immobilized in PMMA transparent films as a selective fluorescence sensor for nitroaromatic explosive vapours. J. Mater. Chem. C 2020, 8, 3626–3630. [Google Scholar] [CrossRef]
- Sousaraei, A.; Queiros, C.; Moscoso, F.G.; Tania, C.; Pedrosa, J.M.; Silva, A.M.G.; Cunha-Silva, L.; Cabanillas-Gonzalez, J. Subppm amine detection via absorption and luminescence turn-on caused by ligand exchange in metal-organic frameworks. Anal. Chem. 2019, 91, 15853–15859. [Google Scholar] [CrossRef] [PubMed]
- Roales, J.; Moscoso, F.G.; Gamez, F.; Lopes-Costa, T.; Sousaraei, A.; Casado, S.; Castro-Smirnov, J.R.; Cabanillas-Gonzalez, J.; Almeida, J.; Queiros, C.; et al. Preparation of luminescent metal-organic framework films by soft-imprinting for 2,4-dinitrotoluene sensing. Materials 2017, 10, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiros, C.; Silva, A.M.G.; de Castro, B.; Cunha-Silva, L. From discrete complexes to metal-organic layered materials: Remarkable hydrogen bonding frameworks. Molecules 2020, 25, 1353. [Google Scholar] [CrossRef] [Green Version]
- Gamonal, A.; Sun, C.; Mariano, A.L.; Fernandez-Bartolom, E.; Guerrero-SanVicente, E.; Vlaisavljevich, B.; Castells-Gil, J.; Marti-Gastaldo, C.; Poloni, R.; Wannemacher, R.; et al. Divergent adsorption-dependent luminescence of amino-functionalized lanthanide metal-organic frameworks for highly sensitive NO2 sensors. J. Phys. Chem. Lett. 2020, 11, 3362–3368. [Google Scholar] [CrossRef]
- Viana, A.M.; Juliao, D.; Mirante, F.; Faria, R.G.; de Castro, B.; Balula, S.S.; Cunha-Silva, L. Straightforward activation of metal-organic framework UiO-66 for oxidative desulfurization processes. Catal. Today 2021, 362, 28–34. [Google Scholar] [CrossRef]
- Viana, A.M.; Ribeiro, S.O.; de Castro, B.; Balula, S.S.; Cunha-Silva, L. Influence of UiO-66(Zr) preparation strategies in its catalytic efficiency for desulfurization process. Materials 2019, 12, 3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granadeiro, C.M.; Ribeiro, S.O.; Karmaoui, M.; Valenca, R.; Ribeiro, J.C.; de Castro, B.; Cunha-Silva, L.; Balula, S.S. Production of ultra-deep sulfur-free diesels using a sustainable catalytic system based on UiO-66(Zr). Chem. Commun. 2015, 51, 13818–13821. [Google Scholar] [CrossRef] [PubMed]
- Sousaraei, A.; Queirós, C.; Moscoso, F.G.; Silva, A.M.G.; Lopes-Costa, T.; Pedrosa, J.M.; Cunha-Silva, L.; Cabanillas-Gonzalez, J. Reversible Protonation of Porphyrinic Metal-Organic Frameworks Embedded in Nanoporous Polydimethylsiloxane for Colorimetric Sensing. Adv. Mater. Interfaces 2021, 2001759. [Google Scholar] [CrossRef]
- Kottke, T.; Stalke, D. Crystal handling at low-temperatures. J. App. Cryst. 1993, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- APEX2. Data Collection Software Version 2012.4; Bruker AXS: Delft, The Netherlands, 2012. [Google Scholar]
- Cryopad. Remote Monit. ControlVersion 1.451Oxf; CryosystemsOxf. Ltd.: Long Hanborough, UK, 2006. [Google Scholar]
- SAINT+. Data Integr. Engine V. 8.27b© 2021–2012; Bruker AXS: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SADABS 2012/1, Bruker AXS Area Detector Scaling and Absorption Correction Program; Bruker AXS: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT-2014, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. SHELXL v. 2014/3, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SmCP1 | TbCP1 | TbCP2 | TbCP3 | |
|---|---|---|---|---|
| Formula | C16H29N2O17Sm | C16H29N2O17Tb | C16H17N2O11Tb | C28H21N4O9Tb |
| Mr | 671.76 | 680.33 | 572.23 | 716.41 |
| Crystal morphology | Colorless plates/prisms | Colorless needles | Pink prisms | Colorless plates |
| Crystal size/mm | 0.18 × 0.07 × 0.05 | 0.19 × 0.05 × 0.04 | 0.33 × 0.10 × 0.08 | 0.19 × 0.06 × 0.04 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n | P21/c | P21/c |
| a/Å | 7.1362(2) | 7.1046(8) | 11.8098(8) | 10.350(2) |
| b/Å | 22.6821(6) | 22.583(2) | 10.7888(7) | 21.289(5) |
| c/Å | 14.2428(4) | 14.2477(16) | 14.3259(8) | 12.490(3) |
| β/° | 92.0350(10) | 92.039(6) | 100.253(2) | 104.125(9) |
| Volume/Å3 | 2303.94(11) | 2284.5(4) | 1796.2(2) | 2668.9(10) |
| Z | 4 | 4 | 4 | 4 |
| ρcalculated/gcm-3 | 1.937 | 1.978 | 2.116 | 2.714 |
| Final R índices [I > 2σ (I)] | R1 = 0.0275 wR2 = 0.0568 | R1 = 0.0494 wR2 = 0.0863 | R1 = 0.0324 wR2 = 0.0611 | R1 = 0.0476 wR2 = 0.0818 |
| R índices (all data) | R1 = 0.0363 wR2 = 0.0611 | R1 = 0.0958 wR2 = 0.1024 | R1 = 0.0400 wR2 = 0.0634 | R1 = 0.0906 wR2 = 0.0960 |
| CP | Equiv | Synthesis | T/°C | t/h | η/% | |||
|---|---|---|---|---|---|---|---|---|
| H2aip | LnCl3.6H2O | NaOH | phen | |||||
| SmCP1 | 1.0 | 0.67 | 1.1 | – | HT | 110 | 72 | 28 |
| 0.64 | 1.2 | MWAS | 2 | 21 | ||||
| TbCP1 | 1.0 | 0.64 | 1.2 * | – | HT | 110 | 72 | 37 |
| 0.67 | 1.1 | MWAS | 6 | 16 | ||||
| TbCP2 | 1.0 | 0.66 | 1.0 | – | HT | 110 | 72 | 27 |
| TbCP3 | 1.0 | 0.66 | 1.2 | 0.59 | HT | 110 | 72 | 47 |
| SmCP1 | TbCP1 | |||||||
|---|---|---|---|---|---|---|---|---|
| D–H···O | d (H···O) | d (D···O) | ∠ (DHO) | D–H···O | d (H···O) | d (D···O) | ∠ (DHO) | |
| O1W–H1W···O5Wiii | 1.927(2) | 2.801(3) | 173(3) | O1W–H1W···O6 | 1.81(3) | 2.662(6) | 156(7) | |
| O1W–H2W···O2 | 1.814(2) | 2.669(3) | 163(3) | O1W–H2W···O9W | 1.84(2) | 2.708(7) | 170(6) | |
| O2W–H3W···O9iv | 1.86(2) | 2.738(3) | 172(3) | O2W–H3W···O4iii | 1.71(3) | 2.603(7) | 167(7) | |
| O2W–H4W···O7i | 1.99(2) | 2.846(3) | 161(3) | O2W–H4W···O9Wiv | 1.86(2) | 2.762(8) | 178(6) | |
| O3W–H5W···O4i | 1.82(2) | 2.675(3) | 160(4) | O3W–H5W···O3iii | 1.80(3) | 2.677(7) | 165(6) | |
| O3W–H6W···O8Wiv | 1.837(2) | 2.722(3) | 175(4) | O3W–H6W···O6W | 1.82(3) | 2.695(7) | 161(6) | |
| O4W–H7W···O8W | 1.870(2) | 2.745(3) | 179(4) | O4W–H7W···O6Wvi | 1.90(4) | 2.726(7) | 153(7) | |
| O4W–H8W···O8v | 1.706(2) | 2.603(3) | 178(4) | O4W–H8W···O3ii | 2.04(4) | 2.846(7) | 149(7) | |
| O5W–H9W···O7v | 1.80(2) | 2.669(3) | 169(3) | O5W–H9W···O7i | 1.79(2) | 2.661(6) | 165(6) | |
| O5W–H10W···O9W | 1.83(2) | 2.693(3) | 167(3) | O5W–H10W···O3Wvii | 1.92(2) | 2.807(7) | 170(6) | |
| O6W–H11W···O8 | 2.49(3) | 3.021(3) | 120(3) | O6W–H11W···O1 | 2.60(5) | 3.124(7) | 117(4) | |
| O6W–H11W···O7Wvi | 2.13(2) | 2.951(4) | 155(3) | O6W–H11W···O2 | 1.83(2) | 2.722(6) | 167(6) | |
| O6W–H12W···O4 | 1.840(2) | 2.712(4) | 171(4) | O6W–H12W···O3v | 1.90(3) | 2.757(6) | 159(7) | |
| O7W–H14W···O3W | 1.88(2) | 2.809(4) | 171(4) | O7W–H13W···O1W | 1.93(2) | 2.833(8) | 169(8) | |
| O8W–H15W···O5 | 2.21(3) | 2.961(3) | 146(3) | O7W–H14W···O6x | 2.47(4) | 3.271(9) | 146(7) | |
| N1–H1···O6vii | 1.96(2) | 2.808(4) | 162(3) | O9W–H17W···O1vi | 2.12(4) | 2.983(7) | 160(7) | |
| N1–H2···O6Wvii | 1.920(2) | 2.797(4) | 176(3) | O9W–H18W···O8W | 1.95(3) | 2.790(8) | 155(7) | |
| N1–H3···O2vii | 1.95(2) | 2.784(4) | 158(3) | O8W–H15W···O4ix | 2.59(9) | 3.025(8) | 124(9) | |
| N2–H10···O6vii | 2.55(3) | 3.174(4) | 130(3) | O8W–H15W···O7W | 2.28(9) | 2.922(9) | 156(11) | |
| O8W–H16W···O6Wviii | 1.92(2) | 2.777(4) | 162(4) | O8W–H16W···O6x | 1.90(8) | 2.707(8) | 172(8) | |
| O9W–H17W···O7vi | 1.881(2) | 2.755(3) | 170(3) | N1–H1···O7W | 2.34(4) | 3.048(9) | 149(7) | |
| O9W–H18W···O5 | 2.56(3) | 3.130(3) | 123(3) | N1–H2···O2ix | 2.54(7) | 3.183(8) | 143(7) | |
| O9W–H18W···O6 | 1.865(2) | 2.742(3) | 172(3) | N2–H2A···O7vii | 2.10 | 2.776(7) | 130 | |
| N2–H9···O7W | 2.28(3) | 3.041(4) | 147(3) | N2–H2B···O8Wii | 2.26 | 2.799(8) | 117 | |
| D–H···O | d (H···O) | d (D···O) | ∠ (DHO) | D–H···O | d (H···O) | d (D···O) | ∠ (DHO) | |
|---|---|---|---|---|---|---|---|---|
| O1W−H2W···O6v | 2.01(4) | 2.759(4) | 164(5) | N1−H3N···O7ix | 2.17(4) | 2.717(4) | 123(4) | |
| O2W−H3W···O8vi | 2.14(2) | 3.024(4) | 167(4) | N2−H4N···O3Wviii | 2.354(19) | 3.193(6) | 171(4) | |
| O2W−H4W···O3W | 1.994(19) | 2.873(5) | 170(4) | N2−H5N···O5viii | 2.26(2) | 3.106(4) | 174(4) | |
| N1−H1N···O2vii | 1.90(3) | 2.719(4) | 154(4) | O3W−H5W···O1 | 2.13(2) | 2.985(5) | 157(5) | |
| N1−H2N···O6viii | 1.80(2) | 2.687(4) | 172(4) |
| D–H···O | d (H···O) | d (D···O) | ∠ (DHO) |
|---|---|---|---|
| N1−H1N···O1Wv | 2.18(4) | 2.984(8) | 156(7) |
| N1−H2N···O1vi | 2.06(3) | 2.888(7) | 162(7) |
| N2−H2A···O1Wvii | 2.36 | 3.085(8) | 136.1 |
| N2−H2C···O8vii | 1.77 | 2.682(3) | 176.9 |
| O1W−H1W···O7 | 1.87(2) | 2.764(7) | 167(7) |
| CP | Synthesis Method | λem/nm | τx/μs (%)(a) | τtotal(b)/µs | X2(c) |
|---|---|---|---|---|---|
| SmCP1 | HT | 590 (max), 483, 493, 518, 560, 565, 605, 642, 649, 698 | 4.43 (100) | 4.43 | 1.015 |
| MWAS | 596 (max), 482, 492, 519, 559, 565, 605, 642, 648, 697 | 4.39 (100) | 4.39 | 1.000 | |
| TbCP1 | HT | 492, 545 (max), 551, 583, 589, 622, 647, 668, 680 | 136.121 (50.14) 43.82 (10.09) 283.94 (39.76) | 185.57 | 1.001 |
| MWAS | 492, 545 (max), 550, 584, 589, 622, 648, 668, 680 | 278.43 (27.95) 121.331 (50.30) 43.662 (21.24) 5.13 (0.50) | 148.15 | 1.004 | |
| TbCP2 | HT | 489, 543 (max), 548, 588, 620, 647 | 137.911 (29.53) 64.737 (57.72) 21.626 (12.76) | 80.85 | 1.049 |
| TbCP3 | HT | 490, 546 (max), 586, 622, 650 | 696.22 (76.77) 0.771 (0.25) 25.186 (15.96) 120.87 (7.02) | 546.99 | 0.975 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queirós, C.; Sun, C.; Silva, A.M.G.; de Castro, B.; Cabanillas-Gonzalez, J.; Cunha-Silva, L. Multidimensional Ln-Aminophthalate Photoluminescent Coordination Polymers. Materials 2021, 14, 1786. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14071786
Queirós C, Sun C, Silva AMG, de Castro B, Cabanillas-Gonzalez J, Cunha-Silva L. Multidimensional Ln-Aminophthalate Photoluminescent Coordination Polymers. Materials. 2021; 14(7):1786. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14071786
Chicago/Turabian StyleQueirós, Carla, Chen Sun, Ana M. G. Silva, Baltazar de Castro, Juan Cabanillas-Gonzalez, and Luís Cunha-Silva. 2021. "Multidimensional Ln-Aminophthalate Photoluminescent Coordination Polymers" Materials 14, no. 7: 1786. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14071786