
Physico-Chemical Modifications Affecting the Activity and Stability of Cu-Based Hybrid Catalysts during the Direct Hydrogenation of Carbon Dioxide into Dimethyl-Ether
 , , , , ,
, , , , ,
Abstract
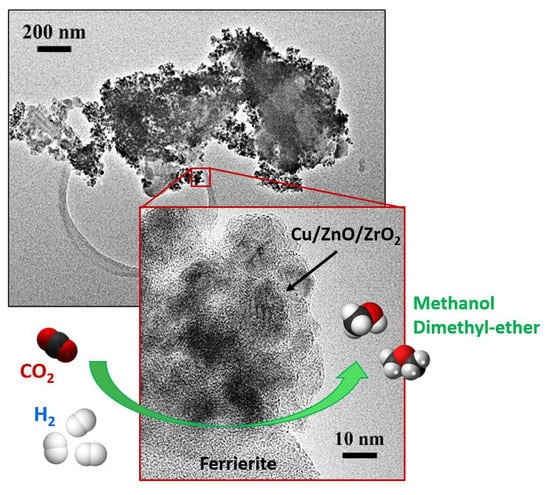
:
1. Introduction
2. Materials and Methods
2.1. Preparation of the Catalytic System
2.2. Characterization of the Catalysts
2.3. Catalytic Tests
3. Results and Discussion
3.1. Physico-Chemical Characterization of the Catalysts
3.1.1. Analytical Composition, Texture, Structure and Morphology
3.1.2. Redox Properties and Acidity
3.2. Catalytic Screening in the Direct Hydrogenation of CO2 to DME
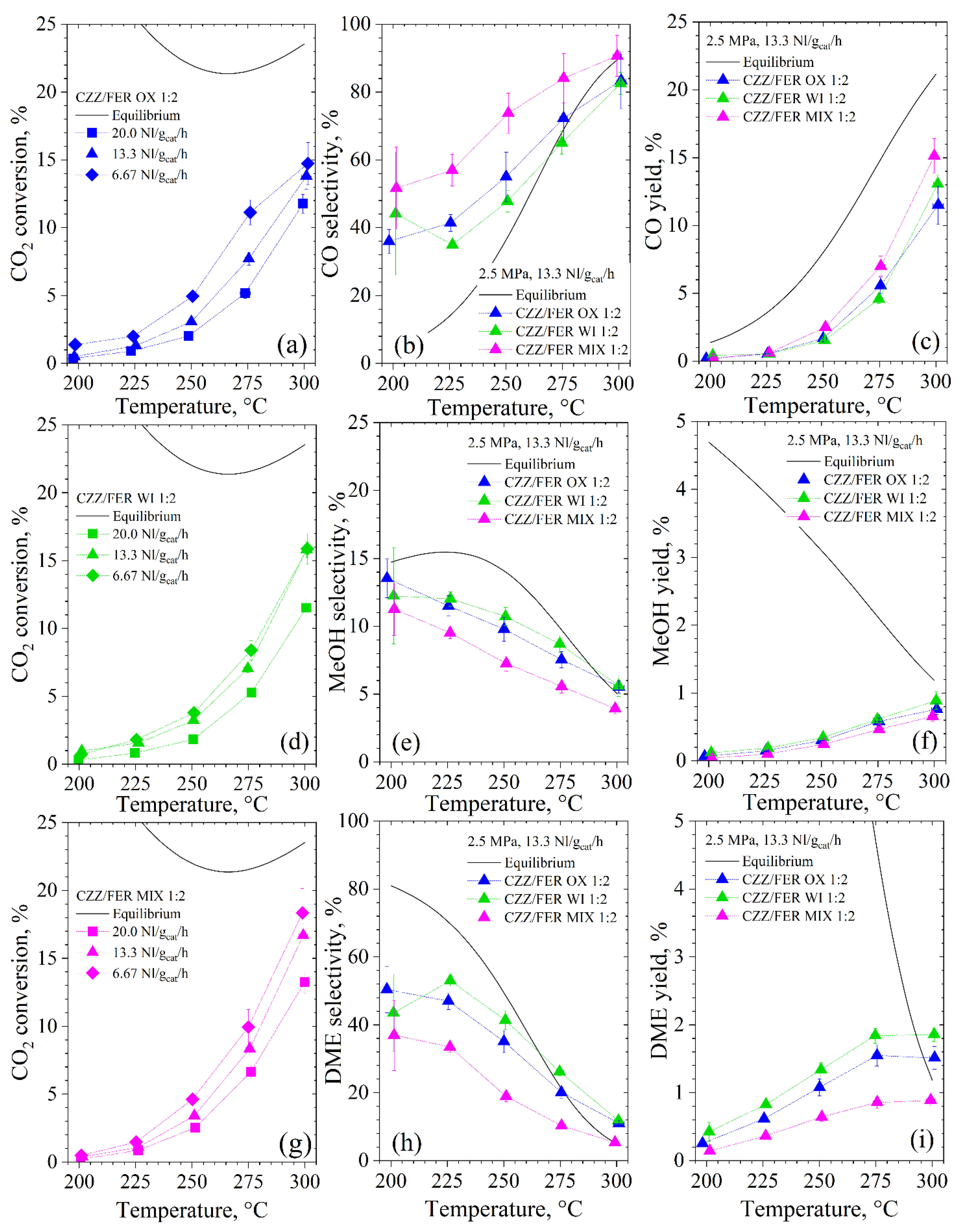
3.2.1. Activity Tests
3.2.2. Structure–Activity Relationships
3.2.3. Stability Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Brynolf, S.; Taljegard, M.; Grahn, M.; Hansson, J. Electrofuels for the Transport Sector: A Review of Production Costs. Renew. Sustain. Energy Rev. 2018, 81, 1887–1905. [Google Scholar] [CrossRef]
- Aneke, M.; Wang, M. Energy Storage Technologies and Real Life Applications—A State of the Art Review. Appl. Energy 2016, 179, 350–377. [Google Scholar] [CrossRef] [Green Version]
- Salomone, F.; Giglio, E.; Ferrero, D.; Santarelli, M.; Pirone, R.; Bensaid, S. Techno-Economic Modelling of a Power-to-Gas System Based on SOEC Electrolysis and CO2 Methanation in a RES-Based Electric Grid. Chem. Eng. J. 2019, 377, 120233. [Google Scholar] [CrossRef]
- Morosanu, E.A.; Salomone, F.; Pirone, R.; Bensaid, S. Insights on a Methanation Catalyst Aging Process: Aging Characterization and Kinetic Study. Catalysts 2020, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Mazza, A.; Salomone, F.; Arrigo, F.; Bensaid, S.; Bompard, E.; Chicco, G. Impact of Power-to-Gas on Distribution Systems with Large Renewable Energy Penetration. Energy Convers. Manag. X 2020, 7, 100053. [Google Scholar] [CrossRef]
- Kar, S.; Kothandaraman, J.; Goeppert, A.; Prakash, G.K.S. Advances in Catalytic Homogeneous Hydrogenation of Carbon Dioxide to Methanol. J. CO2 Util. 2018, 23, 212–218. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Guzmán, H.; Salomone, F.; Batuecas, E.; Tommasi, T.; Russo, N.; Bensaid, S.; Hernández, S. How to Make Sustainable CO2 Conversion to Methanol: Thermocatalytic versus Electrocatalytic Technology. Chem. Eng. J. 2020, 417, 127973. [Google Scholar] [CrossRef]
- Hernández, S. A Review on the Production Processes of Renewable Jet Fuel. Renew. Sustain. Energy Rev. 2017, 79, 709–729. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on Methanation—From Fundamentals to Current Projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl Ether: A Review of Technologies and Production Challenges. Chem. Eng. Process. Process Intensif. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Panzone, C.; Philippe, R.; Chappaz, A.; Fongarland, P.; Bengaouer, A. Power-to-Liquid Catalytic CO2 Valorization into Fuels and Chemicals: Focus on the Fischer-Tropsch Route. J. CO2 Util. 2020, 38, 314–347. [Google Scholar] [CrossRef]
- Buttler, A.; Spliethoff, H. Current Status of Water Electrolysis for Energy Storage, Grid Balancing and Sector Coupling via Power-to-Gas and Power-to-Liquids: A Review. Renew. Sustain. Energy Rev. 2017, 82, 2440–2454. [Google Scholar] [CrossRef]
- Martinez-Burgos, W.J.; de Souza Candeo, E.; Pedroni Medeiros, A.B.; Cesar de Carvalho, J.; Oliveira de Andrade Tanobe, V.; Soccol, C.R.; Sydney, E.B. Hydrogen: Current Advances and Patented Technologies of Its Renewable Production. J. Clean. Prod. 2021, 286, 124970. [Google Scholar] [CrossRef]
- Lepage, T.; Kammoun, M.; Schmetz, Q.; Richel, A. Biomass-to-Hydrogen: A Review of Main Routes Production, Processes Evaluation and Techno-Economical Assessment. Biomass Bioenergy 2021, 144, 105920. [Google Scholar] [CrossRef]
- Godula-Jopek, A.; Stolten, D. Hydrogen Production by Electrolysis; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Zoppi, G.; Pipitone, G.; Pirone, R.; Bensaid, S. Aqueous Phase Reforming Process for the Valorization of Wastewater Streams: Application to Different Industrial Scenarios. Catal. Today 2022, 387, 224–236. [Google Scholar] [CrossRef]
- Zoppi, G.; Pipitone, G.; Gruber, H.; Weber, G.; Reichhold, A.; Pirone, R.; Bensaid, S. Aqueous Phase Reforming of Pilot-Scale Fischer-Tropsch Water Effluent for Sustainable Hydrogen Production. Catal. Today 2021, 367, 239–247. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A Comparative Overview of Hydrogen Production Processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Mezzapica, A.; Frusteri, F. DME Production by CO2 Hydrogenation: Key Factors Affecting the Behaviour of CuZnZr/Ferrierite Catalysts. Catal. Today 2017, 281, 337–344. [Google Scholar] [CrossRef]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to Value-Added Products—A Review and Potential Future Developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Chen, W.-H.; Lin, B.-J.; Lee, H.-M.; Huang, M.-H. One-Step Synthesis of Dimethyl Ether from the Gas Mixture Containing CO2 with High Space Velocity. Appl. Energy 2012, 98, 92–101. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Mezzapica, A.; Spadaro, L.; Arena, F.; Frusteri, F. Catalytic Behaviour of a Bifunctional System for the One Step Synthesis of DME by CO2 Hydrogenation. Catal. Today 2014, 228, 51–57. [Google Scholar] [CrossRef]
- Ali, K.A.; Abdullah, A.Z.; Mohamed, A.R. Recent Development in Catalytic Technologies for Methanol Synthesis from Renewable Sources: A Critical Review. Renew. Sustain. Energy Rev. 2015, 44, 508–518. [Google Scholar] [CrossRef]
- Chen, W.-H.; Hsu, C.-L.; Wang, X.-D. Thermodynamic Approach and Comparison of Two-Step and Single Step DME (Dimethyl Ether) Syntheses with Carbon Dioxide Utilization. Energy 2016, 109, 326–340. [Google Scholar] [CrossRef]
- Bozzano, G.; Manenti, F. Efficient Methanol Synthesis: Perspectives, Technologies and Optimization Strategies. Prog. Energy Combust. Sci. 2016, 56, 71–105. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic Carbon Dioxide Hydrogenation to Methanol: A Review of Recent Studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Sun, K.; Lu, W.; Wang, M.; Xu, X. Low-Temperature Synthesis of DME from CO2/H2 over Pd-Modified CuO-ZnO-Al2O3-ZrO2/HZSM-5 Catalysts. Catal. Commun. 2004, 5, 367–370. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Guo, H.; Tu, X. Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2. ACS Catal. 2018, 8, 90–100. [Google Scholar] [CrossRef]
- Francis, A.; Shanmuga Priya, S.; Harish Kumar, S.; Sudhakar, K.; Tahir, M. A Review on Recent Developments in Solar Photoreactors for Carbon Dioxide Conversion to Fuels. J. CO2 Util. 2021, 47, 101515. [Google Scholar] [CrossRef]
- Guzmán, H.; Salomone, F.; Bensaid, S.; Castellino, M.; Russo, N.; Hernández, S. CO2 Conversion to Alcohols over Cu/ZnO Catalysts: Prospective Synergies between Electrocatalytic and Thermocatalytic Routes. ACS Appl. Mater. Interfaces 2022, 14, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Poto, S.; Gallucci, F.; Fernanda Neira d’Angelo, M. Direct Conversion of CO2 to Dimethyl Ether in a Fixed Bed Membrane Reactor: Influence of Membrane Properties and Process Conditions. Fuel 2021, 302, 121080. [Google Scholar] [CrossRef]
- Ateka, A.; Rodriguez-Vega, P.; Ereña, J.; Aguayo, A.T.; Bilbao, J. Kinetic Modeling and Reactor Design of the Direct Synthesis of Dimethyl Ether for CO2 Valorization. A Review. Fuel 2022, 327, 125148. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, L.; Li, S.; Zhou, Z.; Sun, Y. Novel Heterogeneous Catalysts for CO2 Hydrogenation to Liquid Fuels. ACS Cent. Sci. 2020, 6, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Li, F.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Influence of Modifier (Mn, La, Ce, Zr and Y) on the Performance of Cu/Zn/Al Catalysts via Hydrotalcite-like Precursors for CO2 Hydrogenation to Methanol. Appl. Catal. A Gen. 2013, 468, 442–452. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct Conversion of CO2 into Liquid Fuels with High Selectivity over a Bifunctional Catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Gao, P.; Dang, S.; Li, S.; Bu, X.; Liu, Z.; Qiu, M.; Yang, C.; Wang, H.; Zhong, L.; Han, Y.; et al. Direct Production of Lower Olefins from CO2 Conversion via Bifunctional Catalysis. ACS Catal. 2018, 8, 571–578. [Google Scholar] [CrossRef]
- Frusteri, F.; Migliori, M.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Aloise, A.; Giordano, G.; Bonura, G. Direct CO2 -to-DME Hydrogenation Reaction: New Evidences of a Superior Behaviour of FER-Based Hybrid Systems to Obtain High DME Yield. J. CO2 Util. 2017, 18, 353–361. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives. Molecules 2017, 23, 31. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Spadaro, L.; Cannilla, C.; Arena, F.; Frusteri, F. Hybrid Cu–ZnO–ZrO2/H-ZSM5 System for the Direct Synthesis of DME by CO2 Hydrogenation. Appl. Catal. B 2013, 140–141, 16–24. [Google Scholar] [CrossRef]
- He, F.; Li, Y. Advances on Theory and Experiments of the Energy Applications in Graphdiyne. CCS Chem. 2022, 1–23. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Ge, Q.; Sun, J. Directly Converting CO2 into a Gasoline Fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Jin, B.; Ren, S.; Li, S.; Yu, M.; Liang, X. Roles of Interaction between Components in CZZA/ HZSM-5 Catalyst for Dimethyl Ether Synthesis via CO2 Hydrogenation. AIChE J. 2021, 67, e17353. [Google Scholar] [CrossRef]
- Wild, S.; Polierer, S.; Zevaco, T.A.; Guse, D.; Kind, M.; Pitter, S.; Delgado, K.H.; Sauer, J. Direct DME Synthesis on CZZ/H-FER from Variable CO2/CO Syngas Feeds. RSC Adv. 2021, 11, 2556–2564. [Google Scholar] [CrossRef]
- Ham, H.; Xuan, N.T.; Jung, H.S.; Kim, J.; Roh, H.S.; Bae, J.W. Crucial Factors to Maximize DME Productivity on Hydrophobic Bifunctional Cu-ZnO-Al2O3/Ferrierite by Direct CO2 Hydrogenation. Catal. Today 2021, 369, 112–122. [Google Scholar] [CrossRef]
- Ateka, A.; Portillo, A.; Sánchez-Contador, M.; Bilbao, J.; Aguayo, A.T. Macro-Kinetic Model for CuO–ZnO–ZrO2@SAPO-11 Core-Shell Catalyst in the Direct Synthesis of DME from CO/CO2. Renew. Energy 2021, 169, 1242–1251. [Google Scholar] [CrossRef]
- Chen, H.-B.; Liao, D.-W.; Yu, L.-J.; Lin, Y.-J.; Yi, J.; Zhang, H.-B.; Tsai, K.-R. Influence of Trivalent Metal Ions on the Surface Structure of a Copper-Based Catalyst for Methanol Synthesis. Appl. Surf. Sci. 1999, 147, 85–93. [Google Scholar] [CrossRef]
- Hirano, M.; Akano, T.; Imai, T.; Kuroda, K. Methanol Synthesis from Carbon Dioxide on CuO-ZnO-Al2O3. Energy Convers. Manag. 1995, 36, 585–588. [Google Scholar] [CrossRef]
- Liu, X.-M.; Lu, G.Q.; Yan, Z.-F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- L’hospital, V.; Angelo, L.; Zimmermann, Y.; Parkhomenko, K.; Roger, A.C. Influence of the Zn/Zr Ratio in the Support of a Copper-Based Catalyst for the Synthesis of Methanol from CO2. Catal. Today 2021, 369, 95–104. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The Mechanism of CO and CO2 Hydrogenation to Methanol over Cu-Based Catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Kunkes, E.L.; Studt, F.; Abild-Pedersen, F.; Schlögl, R.; Behrens, M. Hydrogenation of CO2 to Methanol and CO on Cu/ZnO/Al2O3: Is There a Common Intermediate or Not? J. Catal. 2015, 328, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Bahmani, M.; Vasheghani Farahani, B.; Sahebdelfar, S. Preparation of High Performance Nano-Sized Cu/ZnO/Al2O3 Methanol Synthesis Catalyst via Aluminum Hydrous Oxide Sol. Appl. Catal. A Gen. 2016, 520, 178–187. [Google Scholar] [CrossRef]
- Bonura, G.; Migliori, M.; Frusteri, L.; Cannilla, C.; Catizzone, E.; Giordano, G.; Frusteri, F. Acidity Control of Zeolite Functionality on Activity and Stability of Hybrid Catalysts during DME Production via CO2 Hydrogenation. J. CO2 Util. 2018, 24, 398–406. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. The Influence of Zeolite Surface-Aluminum Species on the Deactivation of CuZnAl/Zeolite Hybrid Catalysts for the Direct DME Synthesis. Catal. Today 2014, 227, 144–153. [Google Scholar] [CrossRef]
- Flores, J.H.; Pais da Silva, M.I. Acid properties of the hybrid catalyst CuO–ZnO or CuO–ZnO–Al2O3/H-ferrierite: An infrared study. Colloids Surf. A Physicochem. Eng. Asp. 2008, 322, 113–123. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Giglio, E.; Ferrarelli, G.; Bianco, M.; Migliori, M.; Giordano, G. MFI vs. FER Zeolite during Methanol Dehydration to Dimethyl Ether: The Crystal Size Plays a Key Role. Catal. Commun. 2021, 149, 106214. [Google Scholar] [CrossRef]
- Dai, W.-L.; Sun, Q.; Deng, J.-F.; Wu, D.; Sun, Y.-H. XPS Studies of Cu/ZnO/Al2O3 Ultra-Fine Catalysts Derived by a Novel Gel Oxalate Co-Precipitation for Methanol Synthesis by CO2 + H2. Appl. Surf. Sci. 2001, 177, 172–179. [Google Scholar]
- Sun, Q.; Zhang, Y.-L.; Chen, H.-Y.; Deng, J.-F.; Wu, D.; Chen, S.-Y. A Novel Process for the Preparation of Cu/ZnO and Cu/ZnO/Al2O3 Ultrafine Catalyst: Structure, Surface Properties, and Activity for Methanol Synthesis from CO2 + H2. J. Catal. 1997, 167, 92–105. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The Changing Nature of the Active Site of Cu-Zn-Zr Catalysts for the CO2 Hydrogenation Reaction to Methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent Progress for Direct Synthesis of Dimethyl Ether from Syngas on the Heterogeneous Bifunctional Hybrid Catalysts. Appl. Catal. B 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Bonura, G.; Frusteri, F.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Catalytic Features of CuZnZr–Zeolite Hybrid Systems for the Direct CO2-to-DME Hydrogenation Reaction. Catal. Today 2016, 277, 48–54. [Google Scholar] [CrossRef]
- Langmuir, I. The Adsorption of Gases on Plane Surfaces of Glass, Mica and Platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Harkins, W.D.; Jura, G. Surfaces of Solids. XIII. A Vapor Adsorption of a Monolayer Area, and the Areas Occupied Be Nitrogen and Other Molecules on the Surfase of a Solid. J. Am. Chem. Soc. 1944, 66, 1366–1373. [Google Scholar] [CrossRef]
- De Boer, J.H.; Lippens, B.C.; Linsen, B.G.; Broekhoff, J.C.P.; van den Heuvel, A.; Osinga, T.J. Thet-Curve of Multimolecular N2-Adsorption. J. Colloid Interface Sci. 1966, 21, 405–414. [Google Scholar] [CrossRef]
- Buttersack, C.; Möllmer, J.; Hofmann, J.; Gläser, R. Determination of Micropore Volume and External Surface of Zeolites. Microporous Mesoporous Mater. 2016, 236, 63–70. [Google Scholar] [CrossRef]
- Storck, S.; Bretinger, H.; Maier, W.F. Characterization of Micro- and Mesoporous Solids by Physisorption Methods and Pore-Size Analysis. Appl. Catal. A Gen. 1998, 174, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Condon, J.B. Surface Area and Porosity Determinations by Physisorption Measurements and Theory, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2006; ISBN 9780444519641. [Google Scholar]
- Patterson, A.L. The Scherrer Formula for X-Ray Particle Size Determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Ortega, C.; Rezaei, M.; Hessel, V.; Kolb, G. Methanol to Dimethyl Ether Conversion over a ZSM-5 Catalyst: Intrinsic Kinetic Study on an External Recycle Reactor. Chem. Eng. J. 2018, 347, 741–753. [Google Scholar] [CrossRef]
- Sun, J.T.; Metcalfe, I.S.; Sahibzada, M. Deactivation of Cu/ZnO/Al2O3 Methanol Synthesis Catalyst by Sintering. Ind. Eng. Chem. Res. 1999, 38, 3868–3872. [Google Scholar] [CrossRef]
- Miletto, I.; Catizzone, E.; Bonura, G.; Ivaldi, C.; Migliori, M.; Gianotti, E.; Marchese, L.; Frusteri, F.; Giordano, G. In Situ FT-IR Characterization of CuZnZr/Ferrierite Hybrid Catalysts for One-Pot CO2-to-DME Conversion. Materials 2018, 11, 2275. [Google Scholar] [CrossRef] [Green Version]
- Flores, J.H.; da Silva, M.I.P. Influence of the Preparation Method on Hybrid Catalysts CuO–ZnO–Al2O3 and H-Ferrierite for Syngas Transformation to Hydrocarbons via Methanol. Catal. Lett. 2016, 146, 1505–1516. [Google Scholar] [CrossRef]
- Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Synthesis of DME from Syngas on the Bifunctional Cu–ZnO–Al2O3/Zr-Modified Ferrierite: Effect of Zr Content. Appl. Catal. B 2009, 90, 426–435. [Google Scholar] [CrossRef]
- Wei, P.; Zhu, X.; Wang, Y.; Chu, W.; Xie, S.; Yang, Z.; Liu, X.; Li, X.; Xu, L. Rapid Synthesis of Ferrierite Zeolite through Microwave Assisted Organic Template Free Route. Microporous Mesoporous Mater. 2019, 279, 220–227. [Google Scholar] [CrossRef]
- Mayoral, A.; Anderson, P.A.; Diaz, I. Zeolites Are No Longer a Challenge: Atomic Resolution Data by Aberration-Corrected STEM. Micron 2015, 68, 146–151. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-State Interactions, Adsorption Sites and Functionality of Cu-ZnO/ZrO2 Catalysts in the CO2 Hydrogenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO Synergy in Methanol Synthesis from CO2, Part 2: Origin of the Methanol and CO Selectivities Explained by Experimental Studies and a Sphere Contact Quantification Model in Randomly Packed Binary Mixtures on Cu–ZnO Coprecipitate Catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Frusteri, F. The Influence of Different Promoter Oxides on the Functionality of Hybrid CuZn-Ferrierite Systems for the Production of DME from CO2-H2 Mixtures. Appl. Catal. A Gen. 2017, 544, 21–29. [Google Scholar] [CrossRef]
- García-Trenco, A.; Vidal-Moya, A.; Martínez, A. Study of the Interaction between Components in Hybrid CuZnAl/HZSM-5 Catalysts and Its Impact in the Syngas-to-DME Reaction. Catal. Today 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Dow, W.-P.; Wang, Y.-P.; Huang, T.-J. Yttria-Stabilized Zirconia Supported Copper Oxide Catalyst. J. Catal. 1996, 160, 155–170. [Google Scholar] [CrossRef]
- Grunwaldt, J.-D.; Molenbroek, A.M.; Topsøe, N.-Y.; Topsøe, H.; Clausen, B.S. In Situ Investigations of Structural Changes in Cu/ZnO Catalysts. J. Catal. 2000, 194, 452–460. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol Synthesis. Catal. Today 1992, 15, 51–75. [Google Scholar] [CrossRef]
- Meliàn-Cabrera, I.; Granados, M.L.; Fierro, J.L.G. Pd-Modified Cu?Zn Catalysts for Methanol Synthesis from CO2/H2 Mixtures: Catalytic Structures and Performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Bailey, S.; Waugh, K.C. Comment on the Use of Temperature-Programmed Desorption of H2 as a Tool to Determine Metal Surface Area of Cu Catalysts. Catal. Lett. 1993, 17, 371–374. [Google Scholar] [CrossRef]
- Muhler, M.; Nielsen, L.P.; Törnqvist, E.; Clausen, B.S.; Topsøe, H. Reply to the Comment by Bailey and Waugh on the Use of Temperature Programmed Desorption of H2 to Determine Metal Surface Area of Cu Catalysts. Catal. Lett. 1993, 17, 375–376. [Google Scholar] [CrossRef]
- Ju, W.; Zeng, J.; Bejtka, K.; Ma, H.; Rentsch, D.; Castellino, M.; Sacco, A.; Pirri, C.F.; Battaglia, C. Sn-Decorated Cu for Selective Electrochemical CO2 to CO Conversion: Precision Architecture beyond Composition Design. ACS Appl. Energy Mater. 2019, 2, 867–872. [Google Scholar] [CrossRef]
- Laurenti, M.; Canavese, G.; Stassi, S.; Fontana, M.; Castellino, M.; Pirri, C.F.; Cauda, V. A Porous Nanobranched Structure: An Effective Way to Improve Piezoelectricity in Sputtered ZnO Thin Films. RSC Adv. 2016, 6, 76996–77004. [Google Scholar] [CrossRef]
- Gionco, C.; Hernández, S.; Castellino, M.; Gadhi, T.A.; Muñoz-Tabares, J.A.; Cerrato, E.; Tagliaferro, A.; Russo, N.; Paganini, M.C. Synthesis and Characterization of Ce and Er Doped ZrO2 Nanoparticles as Solar Light Driven Photocatalysts. J. Alloys Compd. 2019, 775, 896–904. [Google Scholar] [CrossRef]
- Hernández, S.; Gionco, C.; Husak, T.; Castellino, M.; Muñoz-Tabares, J.A.; Tolod, K.R.; Giamello, E.; Paganini, M.C.; Russo, N. Insights Into the Sunlight-Driven Water Oxidation by Ce and Er-Doped ZrO2. Front. Chem. 2018, 6, 368. [Google Scholar] [CrossRef] [PubMed]
- Cotirlan, C.; Galca, A.C.; Ciobanu, C.S.; Logofatu, C. The Study of the Silicon Oxide Thickness on Crystalline Si by X-Ray Photoelectron Spectroscopy and Spectroscopic Ellipsometry. J. Optoelectron. Adv. Mater. 2010, 12, 1092–1097. [Google Scholar]
- Mao, D.; Yang, W.; Xia, J.; Zhang, B.; Song, Q.; Chen, Q. Highly Effective Hybrid Catalyst for the Direct Synthesis of Dimethyl Ether from Syngas with Magnesium Oxide-Modified HZSM-5 as a Dehydration Component. J. Catal. 2005, 230, 140–149. [Google Scholar] [CrossRef]
- Kobl, K.; Thomas, S.; Zimmermann, Y.; Parkhomenko, K.; Roger, A.-C. Power-Law Kinetics of Methanol Synthesis from Carbon Dioxide and Hydrogen on Copper–Zinc Oxide Catalysts with Alumina or Zirconia Supports. Catal. Today 2016, 270, 31–42. [Google Scholar] [CrossRef]
- Ateka, A.; Ereña, J.; Bilbao, J.; Aguayo, A.T. Kinetic Modeling of the Direct Synthesis of Dimethyl Ether over a CuO-ZnO-MnO/SAPO-18 Catalyst and Assessment of the CO2 Conversion. Fuel Process. Technol. 2018, 181, 233–243. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | H2 Consumption (mmolH2/gcat) | H2/CuO 1 | Reduced CuO 2 (%) | Reduced ZnO 3 (%) | Reduction Peaks 4 (%) | |||
|---|---|---|---|---|---|---|---|---|
| Tred ≤ 350°C | δ Peak | α | β | γ | ||||
| CuZnZr | 0.94 | 0.28 | 0.13 | 13 | 8 | 10 | 46 | 44 |
| CZZ/FER OX 2:1 | 6.83 | 2.32 | 1.442 | 95 | 98 | 19 | 81 | - |
| CZZ/FER OX 1:2 | 3.67 | 1.18 | 1.42 | ~100 | ~100 | 24 | 62 | 14 |
| CZZ/FER WI 1:2 | 3.80 | 1.18 | 1.60 | 94 | ~100 | 8 | 81 | 11 |
| CZZ-FER MIX 1:2 | 1.95 | 0.51 | 0.82 | 82 | 7 | 12 | 13 | 57 |
| Catalysts | State | NH3 Uptake | Weak Acid Sites 1 | Strong Acid Sites 2 | |
|---|---|---|---|---|---|
| μmolNH3/gcat | μmolNH3/gFER | % | % | ||
| Commercial FER | Fresh reduced | 494 ± 50 | 494 ± 50 | 80 | 20 |
| Spent | - | - | - | - | |
| CZZ/FER OX 1:2 | Fresh reduced | 540 ± 37 | 810 ± 56 | 35 | 65 |
| Spent | 465 ± 12 | 698 ± 18 | 41 | 59 | |
| CZZ/FER WI 1:2 | Fresh reduced | 511 ± 24 | 767 ± 36 | 35 | 65 |
| Spent | 466 ± 17 | 699 ± 26 | 35 | 65 | |
| CZZ-FER MIX 1:2 | Fresh reduced | 270 ± 31 | 405 ± 47 | 60 | 40 |
| Spent | 248 ± 25 | 372 ± 38 | 64 | 36 | |
| CZZ/FER OX 2:1 | Fresh reduced | 271 ± 15 | 813 ± 45 | 42 | 58 |
| Spent | 269 ± 34 | 807 ± 102 | 28 | 72 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomone, F.; Bonura, G.; Frusteri, F.; Castellino, M.; Fontana, M.; Chiodoni, A.M.; Russo, N.; Pirone, R.; Bensaid, S. Physico-Chemical Modifications Affecting the Activity and Stability of Cu-Based Hybrid Catalysts during the Direct Hydrogenation of Carbon Dioxide into Dimethyl-Ether. Materials 2022, 15, 7774. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15217774
Salomone F, Bonura G, Frusteri F, Castellino M, Fontana M, Chiodoni AM, Russo N, Pirone R, Bensaid S. Physico-Chemical Modifications Affecting the Activity and Stability of Cu-Based Hybrid Catalysts during the Direct Hydrogenation of Carbon Dioxide into Dimethyl-Ether. Materials. 2022; 15(21):7774. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15217774
Chicago/Turabian StyleSalomone, Fabio, Giuseppe Bonura, Francesco Frusteri, Micaela Castellino, Marco Fontana, Angelica Monica Chiodoni, Nunzio Russo, Raffaele Pirone, and Samir Bensaid. 2022. "Physico-Chemical Modifications Affecting the Activity and Stability of Cu-Based Hybrid Catalysts during the Direct Hydrogenation of Carbon Dioxide into Dimethyl-Ether" Materials 15, no. 21: 7774. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15217774