
CeFeO3–CeO2–Fe2O3 Systems: Synthesis by Solution Combustion Method and Catalytic Performance in CO2 Hydrogenation
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Synthetic Procedures
2.3. Characterization
2.4. Catalytic Tests
3. Results and Discussion
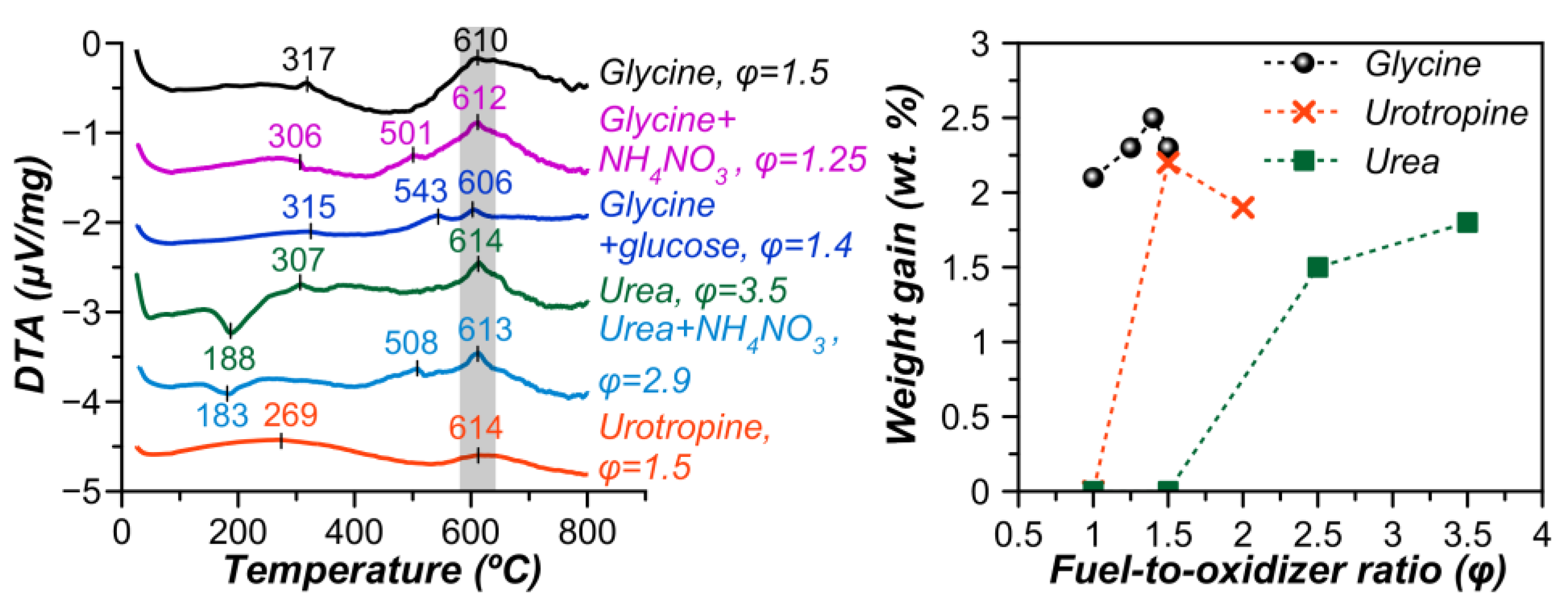
3.1. Synthesis Features and Phase Composition
3.2. Structural and Textural Properties
3.3. Reducibility
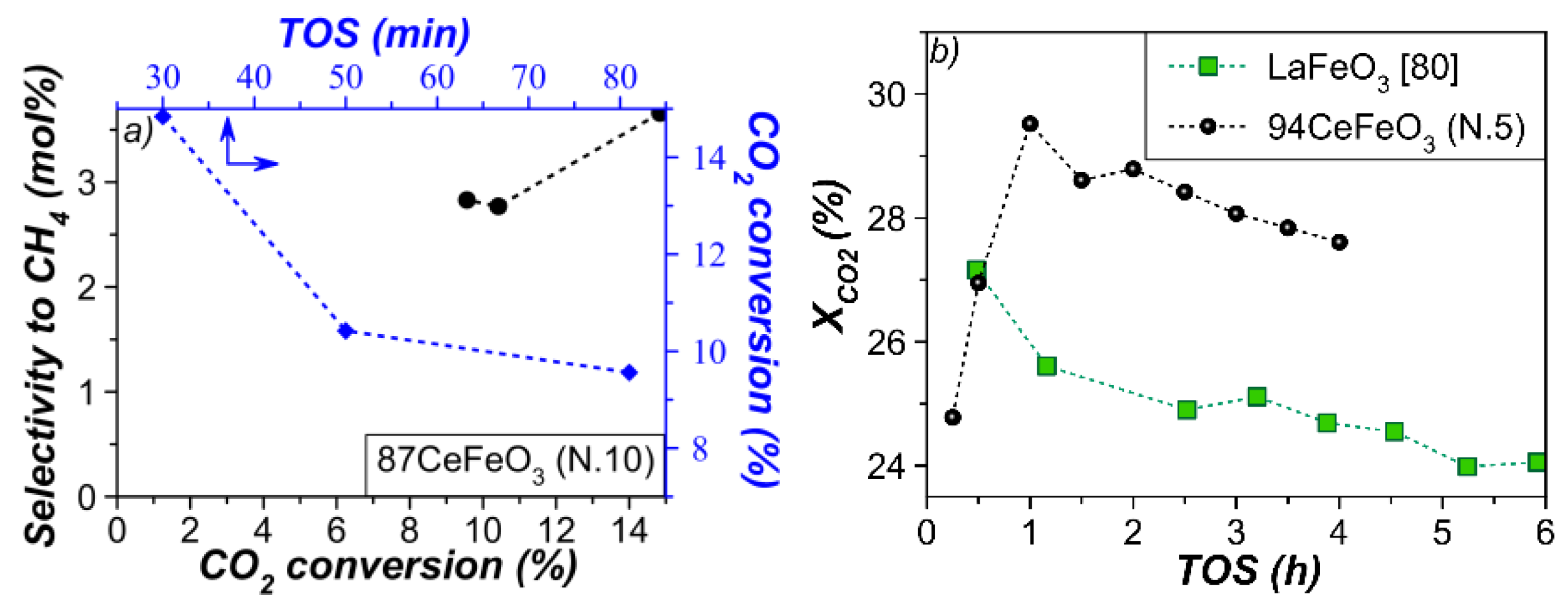
3.4. Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez-Padró, D.; Puente-Santiago, A.R.; Balu, A.M.; Muñ Oz-Batista, M.J.; Luque, R. Environmental Catalysis: Present and Future. ChemCatChem 2019, 11, 18–38. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Review and Evaluation of Hydrogen Production Methods for Better Sustainability. Int. J. Hydrogen Energy 2015, 40, 11094–11111. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen Energy, Economy and Storage: Review and Recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H.; Johnson, D.W.; Remeika, J.P.; Gallagher, P.K. Perovskite Oxides: Materials Science in Catalysis. Science 1977, 195, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Monama, G.R.; Ramohlola, K.E.; Iwuoha, E.I.; Modibane, K.D. Progress on Perovskite Materials for Energy Application. Results Chem. 2022, 4, 100321. [Google Scholar] [CrossRef]
- Yan, S.; Yim, C.-H.; Pankov, V.; Bauer, M.; Baranova, E.; Weck, A.; Merati, A.; Abu-Lebdeh, Y.; Gerbaldi, C. Perovskite Solid-State Electrolytes for Lithium Metal Batteries. Batteries 2021, 7, 75. [Google Scholar] [CrossRef]
- Cao, Y.; Liang, J.; Li, X.; Yue, L.; Liu, Q.; Lu, S.; Asiri, A.M.; Hu, J.; Luo, Y.; Sun, X. Recent Advances in Perovskite Oxides as Electrode Materials for Supercapacitors. Chem. Commun. 2021, 57, 2343–2355. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Meng, H.; Zhu, T.; Yan, D.; Zhu, W.; Liu, S.; Zhao, Q. Perovskite-Perovskite Junctions for Optoelectronics: Fundamentals, Processing, and Applications. Matter 2022, 5, 2086–2118. [Google Scholar] [CrossRef]
- Dang, T.T.; Nguyen, T.L.A.; Ansari, K.B.; Nguyen, V.H.; Binh, N.T.; Phan, T.T.N.; Pham, T.H.; Hang, D.T.T.; Amaniampong, P.N.; Kwao-Boateng, E.; et al. Perovskite Materials as Photocatalysts: Current Status and Future Perspectives. In Nanostructured Photocatalysts: From Fundamental to Practical Applications; Elsevier: Amsterdam, The Netherlands, 2021; pp. 169–216. [Google Scholar] [CrossRef]
- Xu, X.; Pan, Y.; Ge, L.; Shao, Z. Perovskite Materials in Electrocatalysis. In Revolution of Perovskite: Synthesis, Properties and Applications; Arul, N.S., Nithya, V.D., Eds.; Springer: Singapore, 2020; pp. 209–250. ISBN 978-981-15-1267-4. [Google Scholar]
- Roth, R.S. Classification of Perovskite and Other ABO3-Type Compounds. J. Res. Natl. Bur. Stand. 1957, 58, 75–88. [Google Scholar] [CrossRef]
- Bhattar, S.; Abedin, M.A.; Kanitkar, S.; Spivey, J.J. A Review on Dry Reforming of Methane over Perovskite Derived Catalysts. Catal Today 2021, 365, 2–23. [Google Scholar] [CrossRef]
- Joshi, A.; Shah, V.; Mohapatra, P.; Kumar, S.; Joshi, R.K.; Kathe, M.; Qin, L.; Tong, A.; Fan, L.S. Chemical Looping-A Perspective on the next-Gen Technology for Efficient Fossil Fuel Utilization. Adv. Appl. Energy 2021, 3, 100044. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze Der Krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Galasso, F.S. Structure of Perovskite-Type Compounds. In Structure, Properties and Preparation of Perovskite-Type Compounds; Pergamon: Oxford, UK, 1969; pp. 3–49. ISBN 978-0-08-012744-6. [Google Scholar]
- Park, N.G. Perovskite Solar Cells: An Emerging Photovoltaic Technology. Mat. Today 2015, 18, 65–72. [Google Scholar] [CrossRef]
- Vyshatko, N.P.; Kharton, V.; Shaula, A.L.; Naumovich, E.N.; Marques, F.M.B. Structural Characterization of Mixed Conducting Perovskites La(Ga,M)O3−δ (M = Mn, Fe, Co, Ni). Mater. Res. Bull. 2003, 38, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Holtappels, P.; Graule, T.; Nakamura, T.; Gauckler, L.J. Materials Design for Perovskite SOFC Cathodes. Mon. Chem.-Chem. Mon. 2009, 140, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Fedorovskiy, A.E.; Drigo, N.A.; Nazeeruddin, M.K. The Role of Goldschmidt’s Tolerance Factor in the Formation of A2BX6 Double Halide Perovskites and Its Optimal Range. Small Methods 2020, 4, 1900426. [Google Scholar] [CrossRef]
- Inoue, I.H. Electrostatic Carrier Doping to Perovskite Transition-Metal Oxides. Semicond. Sci. Technol. 2005, 20, S112–S120. [Google Scholar] [CrossRef] [Green Version]
- Zaboeva, E.A.; Izotova, S.G.; Popkov, V.I. Glycine-Nitrate Combustion Synthesis of CeFeO3-Based Nanocrystalline Powders. Russ. J. Appl. Chem. 2016, 89, 1228–1236. [Google Scholar] [CrossRef]
- Choong, C.E.; Park, C.M.; Chang, Y.Y.; Yang, J.K.; Kim, J.R.; Oh, S.E.; Jeon, B.H.; Choi, E.H.; Yoon, Y.; Jang, M. Interfacial Coupling Perovskite CeFeO3 on Layered Graphitic Carbon Nitride as a Multifunctional Z-Scheme Photocatalyst for Boosting Nitrogen Fixation and Organic Pollutants Demineralization. Chem. Eng. J. 2022, 427, 131406. [Google Scholar] [CrossRef]
- Bertaut, F.; Forrat, F. Sur Les Déformations Dans Les Pérovskites à Base de Terres Rares et d’éléments de Transition Trivalents. J. Phys. Radium 1956, 17, 129–131. [Google Scholar] [CrossRef]
- Petschnig, L.L.; Fuhrmann, G.; Schildhammer, D.; Tribus, M.; Schottenberger, H.; Huppertz, H. Solution Combustion Synthesis of CeFeO3 under Ambient Atmosphere. Ceram. Int. 2016, 42, 4262–4267. [Google Scholar] [CrossRef]
- Robbins, M.; Wertheim, G.K.; Menth, A.; Sherwood, R.C. Preparation and Properties of Polycrystalline Cerium Orthoferrite (CeFeO3). J. Phys. Chem. Solids 1969, 30, 1823–1825. [Google Scholar] [CrossRef]
- Kitayama, K.; Nojiri, K.; Sugihara, T.; Katsura, T. Phase Equilibria in the CeO and CeFeO Systems. J. Solid State Chem. 1985, 56, 1–11. [Google Scholar] [CrossRef]
- Ameta, J.; Kumar, A.; Ameta, R.; Sharma, V.K.; Ameta, S.C. Synthesis and Characterization of CeFeO3 Photocatalyst Used in Photocatalytic Bleaching of Gentian Violet. J. Iran. Chem. Soc. 2009, 6, 293–299. [Google Scholar] [CrossRef]
- Pandya, H.N.; Kulkarni, R.G.; Parsania, P.H. Study of Cerium Orthoferrite Prepared by Wet Chemical Method. Mater. Res. Bull. 1990, 25, 1073–1077. [Google Scholar] [CrossRef]
- Selvam, K.; Jeeva, S.; Mani, G. Structural, Magnetic and Optical Properties of Cr Doped CeFeO3 Nanoparticles Synthesized by Co-Precipitation Method. JETIR 2019, 6, 951–958. [Google Scholar]
- Surendran, A.; Gupta, N.K.; Aziz, F.; Kushwaha, K.K. Synthesis and Characterization of Ce-Fe Composite Nanoparticles via Sol-Gel Method. J. Nano-Electron. Phys. 2020, 12, 01025. [Google Scholar] [CrossRef]
- Opuchovic, O.; Kreiza, G.; Senvaitiene, J.; Kazlauskas, K.; Beganskiene, A.; Kareiva, A. Sol-Gel Synthesis, Characterization and Application of Selected Sub-Microsized Lanthanide (Ce, Pr, Nd, Tb) Ferrites. Dye. Pigment. 2015, 118, 176–182. [Google Scholar] [CrossRef]
- Matveyeva, A.N.; Omarov, S.O.; Nashchekin, A.V.; Popkov, V.I.; Murzin, D.Y. Catalyst Supports Based on ZnO–ZnAl2O4 Nanocomposites with Enhanced Selectivity and Coking Resistance in Isobutane Dehydrogenation. Dalton Trans. 2022, 51, 12213–12224. [Google Scholar] [CrossRef]
- Omarov, S.O.; Martinson, K.D.; Matveyeva, A.N.; Chebanenko, M.I.; Nevedomskiy, V.N.; Popkov, V.I. Renewable Hydrogen Production via Glycerol Steam Reforming over Ni/CeO2 Catalysts Obtained by Solution Combustion Method: The Effect of Ni Loading. Fuel Process. Technol. 2022, 236, 107429. [Google Scholar] [CrossRef]
- Deganello, F.; Tyagi, A.K. Solution Combustion Synthesis, Energy and Environment: Best Parameters for Better Materials. Prog. Cryst. Growth Charact. Mater. 2018, 64, 23–61. [Google Scholar] [CrossRef]
- Kingsley, J.J.; Patil, K.C. A Novel Combustion Process for the Synthesis of Fine Particle α-Alumina and Related Oxide Materials. Mater. Lett. 1988, 6, 427–432. [Google Scholar] [CrossRef]
- Merzhanov, A.G.; Borovinskaya, I.P. Historical Retrospective of SHS: An Autoreview. Int. J. Self-Propagating High-Temp. Synth. 2008, 17, 242–265. [Google Scholar] [CrossRef]
- Tang, P.; Zhang, J.; Fu, M.; Cao, F.; Lv, C. Characterization and Preparation Nanosized CeFeO3 by a Microwave Process. Integr. Ferroelectr. 2013, 146, 99–104. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 1234, 26, 172–187. [Google Scholar] [CrossRef] [Green Version]
- González-Castaño, M.; Dorneanu, B.; Arellano-García, H. The Reverse Water Gas Shift Reaction: A Process Systems Engineering Perspective. React. Chem. Eng. 2021, 6, 954–976. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 Conversion by Reverse Water Gas Shift Catalysis: Comparison of Catalysts, Mechanisms and Their Consequences for CO2 Conversion to Liquid Fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The Thermodynamics Analysis and Experimental Validation for Complicated Systems in CO2 Hydrogenation Process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- González-Cortés, S.L.; Imbert, F.E. Fundamentals, Properties and Applications of Solid Catalysts Prepared by Solution Combustion Synthesis (SCS). Appl. Catal. A Gen. 2013, 452, 117–131. [Google Scholar] [CrossRef]
- Gheith, M.A. Differential Thermal Analysis of Certain Iron Oxides and Oxide Hydrates. Am. J. Sci. 1952, 250, 677–695. [Google Scholar] [CrossRef]
- Tretyakov, Y.D.; Sorokin, V.V.; Kaul, A.R.; Erastova, A.P. Phase Equilibria and Thermodynamics of Coexisting Phases in Rare-Earth Element-Iron-Oxygen Systems. I. The Cerium-Iron-Oxygen System. J. Solid State Chem. 1976, 18, 253–261. [Google Scholar] [CrossRef]
- Cam, T.S.; Vishnievskaia, T.A.; Popkov, V.I. Catalytic Oxidation of CO over CuO/CeO2 Nanocomposites Synthesized via Solution Combustion Method: Effect of Fuels. Rev. Adv. Mater. Sci. 2020, 59, 131–143. [Google Scholar] [CrossRef]
- Novitskaya, E.; Kelly, J.P.; Bhaduri, S.; Graeve, O.A. A Review of Solution Combustion Synthesis: An Analysis of Parameters Controlling Powder Characteristics. Int. Mater. Rev. 2021, 66, 188–214. [Google Scholar] [CrossRef]
- Pourgolmohammad, B.; Masoudpanah, S.M.; Aboutalebi, M.R. Effects of the Fuel Type and Fuel Content on the Specific Surface Area and Magnetic Properties of Solution Combusted CoFe2O4 Nanoparticles. Ceram. Int. 2017, 43, 8262–8268. [Google Scholar] [CrossRef]
- Varma, A.; Mukasyan, A.S.; Rogachev, A.S.; Manukyan, K.V. Solution Combustion Synthesis of Nanoscale Materials. Chem. Rev. 2016, 116, 14493–14586. [Google Scholar] [CrossRef] [PubMed]
- Civera, A.; Pavese, M.; Saracco, G.; Specchia, V. Combustion Synthesis of Perovskite-Type Catalysts for Natural Gas Combustion. Catal. Today 2003, 83, 199–211. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Bliznuk, V.; Detavernier, C.; Marin, G.B. CeO2-Modified Fe2O3 for CO2 Utilization via Chemical Looping. Ind. Eng. Chem. Res. 2013, 52, 8416–8426. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Kang, W.; Xie, K. Comparison of Reduction Behavior of Fe2O3, ZnO and ZnFe2O4 by TPR Technique. J. Nat. Gas Chem. 2009, 18, 110–113. [Google Scholar] [CrossRef]
- Wei, X.; Zhou, Y.; Li, Y.; Shen, W. Polymorphous Transformation of Rod-Shaped Iron Oxides and Their Catalytic Properties in Selective Reduction of NO by NH3. RSC Adv. 2015, 5, 66141–66146. [Google Scholar] [CrossRef]
- Saharuddin, T.S.; Samsuri, A.; Salleh, F.; Othaman, R.; Kassim, M.; Hisham, M.W.; Yarmo, M.A. The Reduction Behaviour of Cerium Doped Iron Oxide in Hydrogen and Carbon Monoxide Atmosphere. In Materials Science Forum; Trans Tech Publications Ltd.: Wallerau, Switzerland, 2016; Volume 840, pp. 381–385. [Google Scholar]
- Perez-Alonso, F.J.; Melián-Cabrera, I.; López Granados, M.; Kapteijn, F.; Fierro, J.L.G. Synergy of FexCe1−xO2 Mixed Oxides for N2O Decomposition. J. Catal. 2006, 239, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Mutter, D.; Schierholz, R.; Urban, D.F.; Heuer, S.A.; Ohlerth, T.; Kungl, H.; Elsässer, C.; Eichel, R.-A. Defects and phase formation in non-stoichiometric LaFeO3: A combined theoretical and experimental study. Chem. Mater. 2021, 33, 9473–9485. [Google Scholar] [CrossRef]
- Mihai, O.; Chen, D.; Holmen, A. Chemical Looping Methane Partial Oxidation: The Effect of the Crystal Size and O Content of LaFeO3. J. Catal. 2012, 293, 175–185. [Google Scholar] [CrossRef]
- Faye, J.; Baylet, A.; Trentesaux, M.; Royer, S.; Dumeignil, F.; Duprez, D.; Valange, S.; Tatibouët, J.M. Influence of Lanthanum Stoichiometry in La1−xFeO3−δ Perovskites on Their Structure and Catalytic Performance in CH4 Total Oxidation. Appl. Catal. B Env. 2012, 126, 134–143. [Google Scholar] [CrossRef]
- Heidinger, B.; Royer, S.; Alamdari, H.; Giraudon, J.-M.; Lamonier, J.-F. Reactive Grinding Synthesis of LaBO3 (B: Mn, Fe) Perovskite; Properties for Toluene Total Oxidation. Catalysts 2019, 9, 633. [Google Scholar] [CrossRef] [Green Version]
- Qiao, D.; Lu, G.; Liu, X.; Guo, Y.; Wang, Y.; Guo, Y. Preparation of Ce12xFexO2 Solid Solution and Its Catalytic Performance for Oxidation of CH4 and CO. J. Mat. Sci. 2011, 46, 3500–3506. [Google Scholar] [CrossRef]
- He, J.; Zhang, H.; Wang, W.; Yao, P.; Jiao, Y.; Wang, J.; Chen, Y. Soot Combustion over CeO2 Catalyst: The Influence of Biodiesel Impurities (Na, K, Ca, P) on Surface Chemical Properties. Environ. Sci. Pollut. Res. 2021, 28, 26018–26029. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Su, D.; Wang, Z.; Chang, J.; Ma, C. NO Reduction by CO over Copper Catalyst Supported on Mixed CeO2 and Fe2O3: Catalyst Design and Activity Test. Appl. Catal. B 2018, 239, 485–501. [Google Scholar] [CrossRef]
- Spencer, M.S. On the Activation Energies of the Forward and Reverse Water-Gas Shift Reaction. Cat. Lett. 1995, 32, 9–13. [Google Scholar] [CrossRef]
- Landau, M.V.; Meiri, N.; Utsis, N.; Vidruk Nehemya, R.; Herskowitz, M. Conversion of CO2, CO, and H2 in CO2 Hydrogenation to Fungible Liquid Fuels on Fe-Based Catalysts. Ind. Eng. Chem. Res. 2017, 56, 13334–13355. [Google Scholar] [CrossRef]
- Utsis, N.; Vidruk-Nehemya, R.; Landau, M.V.; Herskowitz, M. Novel Bifunctional Catalysts Based on Crystalline Multi-Oxide Matrices Containing Iron Ions for CO2 Hydrogenation to Liquid Fuels and Chemicals. Faraday Discuss. 2016, 188, 545–563. [Google Scholar] [CrossRef]
- Li, S.; Ding, W.; Meitzner, G.D.; Iglesia, E. Spectroscopic and Transient Kinetic Studies of Site Requirements in Iron-Catalyzed Fischer−Tropsch Synthesis. J. Phys. Chem. B 2001, 106, 85–91. [Google Scholar] [CrossRef] [Green Version]
- de Smit, E.; Weckhuysen, B.M. The Renaissance of Iron-Based Fischer-Tropsch Synthesis: On the Multifaceted Catalyst Deactivation Behaviour. Chem. Soc. Rev. 2008, 37, 2758–2781. [Google Scholar] [CrossRef]
- Wezendonk, T.A.; Sun, X.; Dugulan, A.I.; van Hoof, A.J.F.; Hensen, E.J.M.; Kapteijn, F.; Gascon, J. Controlled Formation of Iron Carbides and Their Performance in Fischer-Tropsch Synthesis. J. Catal. 2018, 362, 106–117. [Google Scholar] [CrossRef]
- Riedel, T.; Claeys, M.; Schulz, H.; Schaub, G.; Nam, S.S.; Jun, K.W.; Choi, M.J.; Kishan, G.; Lee, K.W. Comparative Study of Fischer–Tropsch Synthesis with H2/CO and H2/CO2 Syngas Using Fe- and Co-Based Catalysts. Appl. Catal. A Gen. 1999, 186, 201–213. [Google Scholar] [CrossRef]
- Landau, M.V.; Vidruk, R.; Herskowitz, M. Sustainable Production of Green Feed from Carbon Dioxide and Hydrogen. ChemSusChem 2014, 7, 785–794. [Google Scholar] [CrossRef]
- Cui, X.; Xu, J.; Zhang, C.; Yang, Y.; Gao, P.; Wu, B.; Li, Y. Effect of Pretreatment on Precipitated Fe–Mo Fischer–Tropsch Catalysts: Morphology, Carburization, and Catalytic Performance. J. Catal. 2011, 282, 35–46. [Google Scholar] [CrossRef]
- Ding, M.; Yang, Y.; Wu, B.; Wang, T.; Ma, L.; Xiang, H.; Li, Y. Transformation of Carbonaceous Species and Its Influence on Catalytic Performance for Iron-Based Fischer–Tropsch Synthesis Catalyst. J. Mol. Catal. A Chem. 2011, 351, 165–173. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Song, Y.; Song, C.; Guo, X. Selective Hydrogenation of CO2 to Hydrocarbons: Effects of Fe3O4 Particle Size on Reduction, Carburization, and Catalytic Performance. Energy Fuels 2021, 35, 10703–10709. [Google Scholar] [CrossRef]
- De la Rosa-Priego, F.A.; Gutierrez-López, E.D.; Zepeda, T.A.; Acosta-Alejandro, M.; Venezia, A.M.; Fuentes-Moyado, S.; Pawelec, B.; Noé Díaz-de-León, J. Enhanced CO2 Hydrogenation to C2+ Hydrocarbons over Mesoporous X%Fe2O3–Al2O3 Catalysts. Ind. Eng. Chem. Res. 2021, 60, 18660–18671. [Google Scholar] [CrossRef]
- Pérez-Alonso, F.J.; López Granados, M.; Ojeda, M.; Terreros, P.; Rojas, S.; Herranz, T.; Fierro, J.L.G.; Gracia, M.; Gancedo, J.R. Chemical Structures of Coprecipitated Fe−Ce Mixed Oxides. Chem. Mater. 2005, 17, 2329–2339. [Google Scholar] [CrossRef]
- Pathak, L.C.; Singh, T.B.; Das, S.; Verma, A.K.; Ramachandrarao, P. Effect of PH on the Combustion Synthesis of Nano-Crystalline Alumina Powder. Mater. Lett. 2002, 57, 380–385. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, J.S.; Shin, W.C.; Choi, M.J.; Choung, S.J. Catalyst Deactivation during Hydrogenation of Carbon Dioxide: Effect of Catalyst Position in the Packed Bed Reactor. J. Mol. Catal. A Chem. 2009, 301, 98–105. [Google Scholar] [CrossRef]
- Albrecht, M.; Rodemerck, U.; Schneider, M.; Bröring, M.; Baabe, D.; Kondratenko, E.V. Unexpectedly Efficient CO2 Hydrogenation to Higher Hydrocarbons over Non-Doped Fe2O3. Appl. Catal. B 2017, 204, 119–126. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Tian, J.; Yan, B. Cu/LaFeO3 as an Efficient and Stable Catalyst for CO2 Reduction: Exploring Synergistic Effect between Cu and LaFeO3. AIChE J. 2022, 68, e17640. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Héroguel, F.; Kılıç, M.; Baranowski, C.J.; Artiglia, L.; Röthlisberger, U.; Luterbacher, J.S.; Kröcher, O. Cu–Al Spinel as a Highly Active and Stable Catalyst for the Reverse Water Gas Shift Reaction. ACS Catal. 2019, 9, 6243–6251. [Google Scholar] [CrossRef]
- Wei, G.; Zhou, H.; Huang, Z.; Zheng, A.; Zhao, K.; Lin, Y.; Chang, G.; Zhao, Z.; Li, H.; Fang, Y. Reaction Performance of Ce-Enhanced Hematite Oxygen Carrier in Chemical Looping Reforming of Biomass Pyrolyzed Gas Coupled with CO2 Splitting. Energy 2021, 215, 119044. [Google Scholar] [CrossRef]
- Li, K.; Haneda, M.; Ning, P.; Wang, H.; Ozawa, M. Microstructure and Oxygen Evolution of Fe–Ce Mixed Oxides by Redox Treatment. Appl. Surf. Sci. 2014, 289, 378–383. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1101. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Study of Iron-Promoted Cu/SiO2 Catalyst on High Temperature Reverse Water Gas Shift Reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Rui, N.; Han, L.; Zhang, F.; Gerlak, C.A.; Liu, Z.; Cen, J.; Song, L.; Senanayake, S.D.; et al. Conversion of CO2 on a Highly Active and Stable Cu/FeOx/CeO2 Catalyst: Tuning Catalytic Performance by Oxide-Oxide Interactions. Catal. Sci. Technol. 2019, 9, 3735–3742. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample N | Fuel | n(AN)/ n(MeN) | φ | Phase Composition According Rietveld/Corrected 1, wt% | D (1), nm | D (2), nm | D (3), nm | Rwp/Re | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CeFeO3 | CeO2 | Fe2O3 | ||||||||
| 1 | Glycine | – | 1 | 81/74 | 19/18 | 0/8 | 67.5 | 4.7 | – | – |
| 2 | Glycine | – | 1.25 | 78 | 12 | 10 | 66.0 | 7.5 | 6.7 | 1.14 |
| 3 | Glycine | – | 1.4 | 95/93 | 5/5 | 0/2 | 61.3 | 25.0 | – | 1.16 |
| 4 | Glycine | – | 1.5 | 87/82 | 13/12 | 0/6 | 56.8 | 9.0 | – | – |
| 5 | Glycine | 1.5 | 1.25 | 96/94 | 4/4 | 0/2 | 69.1 | 15.6 | – | 1.13 |
| 6 | Glycine | 1.5 | 1.75 | 33 | 47 | 20 | 32.7 | 14.8 | 14.7 | 1.03 |
| 7 | Glycine + glucose | – | 1.4 | 63 | 23 | 14 | 52.0 | 19.4 | 14.3 | – |
| 8 | Urea | – | 2.5 | 52 | 28 | 20 | 68.6 | 5.8 | 4.0 | – |
| 9 | Urea | – | 3.5 | 80 | 13 | 7 | 72.0 | 4.7 | 6.3 | 1.09 |
| 10 | Urea | 1.5 | 2.9 | 91/87 | 9/7 | 0/4 | 66.5 | 22.0 | – | 1.17 |
| 11 | Urotropine | – | 1.5 | 48 | 33 | 19 | 75.1 | 22.1 | 15.8 | – |
| 12 | Urotropine | – | 2 | 61 | 23 | 16 | 60.0 | 16.4 | 7.1 | 1.09 |
| Sample N. | Fuel | φ | D (CeO2), nm | Total Weight Loss According to DTA, wt% |
|---|---|---|---|---|
| 13 | Glycine + glucose | 2.2 | 1.9 | 29.6 |
| 14 | Urea | 1 | 5.7 | 4.6 |
| 15 | Urea | 1.5 | 5.0 | 4.4 |
| 16 | Urotropine | 1 | 6.5 | 5.5 |
| 17 | Urotropine | 6 | 4.7 | – |
| Sample N. | Fuel, Additive, φ | Mass Fraction of CeFeO3 | SBET, m2/g | ΣVpore, cm3/g | Average Pore Size, nm |
|---|---|---|---|---|---|
| 3 | Glycine, 1.4 | 0.93 | 3 | 0.01 | 16.5 |
| 5 | Glycine, NH4NO3, 1.25 | 0.94 | 2 | 0.01 | 15.0 |
| 7 | Glycine, glucose, 1.4 | 0.63 | 8 | 0.12 | 60.5 |
| 9 | Urea, 3.5 | 0.80 | 3 | 0.05 | 59.0 |
| 12 | Urotropine, 2 | 0.61 | 4 | 0.05 | 47.5 |
| 15 | Urea, 1.5 | 0 | 81 | 0.13 | 6.3 |
| Sample | Unit-Cell Volume (CeFeO3), Å3 | Lattice Parameter (CeO2), Å | Theoretical H2 Uptake 1, mmol/g | H2 Uptake According to TPR, mmol/g | |||||
|---|---|---|---|---|---|---|---|---|---|
| CeO2 | Fe2O3 | Total | Area 1 (α + ε) | Area 2 (β + δ) | Area 3 (γ + ζ) | Total | |||
| N.15 | – | 5.376 | 2.05 | 6.15 | 8.20 | 3.77 | 5.42 | 0.7 | 9.89 |
| N.6 | 240.120 | 5.406 | 1.37 | 3.76 | 5.12 | 1.37 | 4.49 | 1.98 | 7.84 |
| N.11 | 239.833 | 5.408 | 0.96 | 3.57 | 4.53 | 1.13 | 2.82 | 2.82 | 6.77 |
| N.4 | 239.511 | 5.402 | 0.35 | 1.13 | 1.48 | 0.73 | 1.29 | 5.23 | 7.25 |
| N.5 | 239.930 | 5.396 | 0.12 | 0.38 | 0.49 | 0.62 | 0.78 | 5.69 | 7.09 |
| Catalyst | H2:CO2 | WHSV, mL·g–1·h–1 | CO Selectivity, % | Rate (mmol CO2/(gcat·s)) | Ref. |
|---|---|---|---|---|---|
| 94CeFeO3–CeO2–Fe2O3 | 1:1 | 72,000 | 100 | 0.064 | This work |
| 5Cu/LaFeO3 | 1:1 | 72,000 | 100 | 0.10 | [80] |
| Commercial Cu/ZnO/Al2O3 | 2:1 | 300,000 | 100 | 0.076 | [81] |
| CuAl2O4 | 2:1 | 300,000 | 100 | 0.005 | [81] |
| Cu−Fe/SiO2 | 1:1 | 120,000 | no data | 0.037 | [86] |
| 5Fe/CeO2 | 1:1 | 600,000 | no data | 0.389 | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matveyeva, A.N.; Omarov, S.O.; Gavrilova, M.A.; Sladkovskiy, D.A.; Murzin, D.Y. CeFeO3–CeO2–Fe2O3 Systems: Synthesis by Solution Combustion Method and Catalytic Performance in CO2 Hydrogenation. Materials 2022, 15, 7970. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15227970
Matveyeva AN, Omarov SO, Gavrilova MA, Sladkovskiy DA, Murzin DY. CeFeO3–CeO2–Fe2O3 Systems: Synthesis by Solution Combustion Method and Catalytic Performance in CO2 Hydrogenation. Materials. 2022; 15(22):7970. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15227970
Chicago/Turabian StyleMatveyeva, Anna N., Shamil O. Omarov, Marianna A. Gavrilova, Dmitry A. Sladkovskiy, and Dmitry Yu. Murzin. 2022. "CeFeO3–CeO2–Fe2O3 Systems: Synthesis by Solution Combustion Method and Catalytic Performance in CO2 Hydrogenation" Materials 15, no. 22: 7970. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15227970