
The Preparation, Characterization, and Pressure-Influenced Dihydrogen Interactions of Tetramethylphosphonium Borohydride

Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-089 Warsaw, Poland
Materials 2023, 16(15), 5334; https://0-doi-org.brum.beds.ac.uk/10.3390/ma16155334
Submission received: 24 June 2023
/
Revised: 22 July 2023
/
Accepted: 25 July 2023
/
Published: 29 July 2023
(This article belongs to the Special Issue Hydrogen Storage in Metal Hydrides and Related Materials)
Abstract
:Tetramethylphosphonium borohydride was synthesized via an ion metathesis reaction in a weakly-coordinating aprotic environment. [(CH3)4P]BH4, in contrast to related [(CH3)4N]+ compounds which tend to crystallize in a tetragonal system, adopts the distorted wurtzite structure (P63mc), resembling some salts containing analogous ions of As and Sb. [(CH3)4P]BH4 decomposes thermally in several endo- and exothermic steps above ca. 240 °C. This renders it more stable than [(CH3)4N]BH4, with a lowered temperature of decomposition onset by ca. 20 °C and solely exothermic processes observed. Raman spectra measured at the 0–10 GPa range indicate that a polymorphic transition occurs within 0.53–1.86 GPa, which is further confirmed by the periodic DFT calculations. The latter suggests a phase transition around 0.8 GPa to a high-pressure phase of [(CH3)4N]BH4. The P63mc phase seems to be destabilized under high pressure by relatively closer dihydrogen interactions, including the C–H…H–C contacts.
1. Introduction
In about the last two decades, borohydrides have become one of the most intensely investigated groups of materials [1,2]. This has been motivated mainly by a high gravimetric content of hydrogen in these compounds, rendering some of them prospective for onboard hydrogen storage for fuel cell vehicles. Obviously, high hydrogen content alone (>6.5 wt%) is not sufficient for a material to fulfill the economically-based requirements listed by the U.S. Department of Energy [3]. Besides their minimal capacity, the other parameters also remain crucial for potential applicability, especially the temperature of H2 evolution (preferably not very far from 100 °C) and the high purity of the released gas.
In order to meet the required parameters of hydrogen storage, various simple and complex borohydrides have been screened, stimulating the development of synthetic capabilities [4]. In this context, borohydrides based on organic cations have received significant interest as precursors for the synthesis of various borohydride complexes [5,6,7,8]. Such organic borohydrides, typically applied as reducing agents [9], are soluble in a wide variety of solvents, which facilitates their use in salt metathesis reactions.
Tetraorgano-phosphonium compounds tend to be more thermally stable in comparison to their ammonium counterparts [10], which may be useful for the stabilization of synthetic precursors [5] or in other potential applications. On the other hand, the hydrogen atoms closest to the phosphorous atom are significantly more acidic in tetraalkylphosphonium cations than those in their ammonium analogues [11]. It would be interesting to establish whether this feature could promote the hydrogen evolution from tetraalkylphosphonium borohydrides under elevated temperatures due to interaction of hydridic (B–Hδ−) and weakly protic (P–C–Hδ+) hydrogen atoms. Significantly elevated pressure, bringing the B–H…H–C distances expected for the solid-state structure of these materials closer together, could reveal a similar destabilizing effect. Such tetraalkylphosphonium borohydrides have not been reported before, contrary to tetraphenylphosphonium borohydride, which has already been utilized for the preparation of numerous borohydride complexes [5,6,8,12,13,14].
To elucidate their structural features and to check their stability under high temperatures or pressurization, we focused on the simplest compound, tetramethylphosphonium borohydride, [(CH3)4P]BH4. We prepared this material and characterized it thoroughly using several analytical techniques, allowing for confirmation of its identity and purity [15,16,17]. We discuss its crystal structure and thermal decomposition and compare the high-pressure behavior of this compound to its lighter analogue, [(CH3)4N]BH4. This is achieved by Raman spectroscopy measurements for the sample compressed in a diamond anvil cell to >10 GPa and theoretical calculations utilizing DFT formalism for periodic systems.
2. Materials and Methods
All of the manipulations were performed in an inert gas (Ar) atmosphere, either in Schlenk-type glassware or in a glovebox (<1 ppm O2, <1 ppm H2O); the latter was also used for storage of the reagents and products. The synthetic procedures are described below.
2.1. Mechanochemical Attempt at Synthesis
[(CH3)4P]Br, 3 mmol, and LiBH4, 3.06 mmol, 102% (both Sigma-Aldrich, St. Louis, MO, USA) were weighted, mixed, and placed in a stainless steel vessel sealed under Ar. A Testchem laboratory vibrational mill (LMW-S) was used for milling, with the nominal frequency of 1400 rpm. The mechanochemical reaction was carried out for 40 min in 5-min periods alternated with several minutes rest to avoid overheating and thermal decomposition of the products. The vessel was cooled with liquid nitrogen to maintain it at room temperature.
2.2. Synthesis of [(CH3)4P][Al(OC(CF3)3)4]
The mixture of water and acetone (ca. 2:1) was used as a solvent, similar to the ref. [18]. A solution of 1.72 mmol of (CH3)4PBr in 9 mL of this solvent was added to the solution of 1.72 mmol of Li[Al(OC(CF3)3)4] (synthesized according to ref. [19]) in ca. 27 mL of the solvent. The mixture was stirred in an open container for ca. 24 h at 40 °C to allow for slow evaporation of the acetone and crystallization of the product poorly soluble in water (isolated with ca. 80% yield).
2.3. Solvent-Mediated Synthesis of [(CH3)4P]BH4
The reaction was performed using an H-vessel equipped with a glass frit (no. 4) [20]. Approximately 0.84 mmol (105%) of [(n-C4H9)4N]BH4 dissolved in 5 mL dichloromethane (DCM) was added to 0.8 mmol of [(CH3)4P][Al(OC(CF3)3)4] suspended in DCM (12 mL). The mixture was stirred for 30 min while the low-density product gathered on the top of the solution. The by-product, [(n-C4H9)4N][Al(OC(CF3)3)4], was extracted using five cycles of filtration and distillation, followed by evaporation of the solvent.
2.4. Analytical Techniques and Data Processing
Single crystals of [(CH3)4P]BH4 were grown from anhydrous acetonitrile solution via slow evaporation of the solvent. The crystals were covered with Krytox 1531 oil and mounted on a goniometer using a nylon loop. The measurements of single-crystal diffraction were performed using an Agilent Supernova X-ray diffractometer equipped with a CuKα radiation micro-source. CrysAlisPro software (v. 38.43) [21] was used for data reduction, while SHELXT [22] and Olex2 [23] were utilized for the structure solution and refinement, respectively. The B–H and C–H were restrained at 1.190(10) Å and 1.083(10) Å, respectively, and all of the hydrogen atoms were refined with fixed displacement parameters, [U(H) = 1.5Ueq(C or B)]. For the structure measured at 300 K and for [(CH3)4P]Cl as well, several DANG restraints involving hydrogen atoms were applied. An Agilent Supernova X-ray diffractometer (CuKα radiation micro-source) and Panalytical X’Pert Pro diffractometer (parallel beam; CoKα radiation) were used to measure the powder diffraction patterns (PXDs). Jana2020 [24] was utilized for Rietveld refinement of the sample of [(CH3)4P]BH4 powder. If not stated otherwise, all of the geometrical parameters mentioned in the text are related to the single-crystal structures measured at 100 K due to their best achievable quality.
CCDC entries 2271774–2271777 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Raman spectra were measured using a Horiba T64000 Raman spectrometer from Jobin Yvon equipped with a Si CCD detector. A laser of 532 nm wavelength was used, and the spectra were collected at room temperature under a microscope using a 10× objective. The samples were sealed in 1 mm Ar-filled quartz capillaries or placed in a diamond anvil cell (DAC). A Diacell® One20DAC from Almax (Diksmuide, Belgium) equipped with type IIa Boehler-Almax diamond Raman-grade anvils with 500 μm culets was used. Stainless steel gaskets were used with a hole of ca. 250 μm drilled after indentation. Krytox 1531 oil was used as a pressure-transmitting medium, and the crystals of the samples were loaded together with the small scraps of ruby necessary for pressure calibration.
TGA/DSC measurements were performed using a Netzsch STA 409 PG instrument. The samples of ca. 10 mg were heated from 20 °C to 450 °C (10 °C min−1 heating rate) in Al2O3 crucibles under Ar flow (100 mL min−1). The results were processed using Netzsch Proteus® software v. 4.8.4. The onset decomposition temperature was estimated as an intersection of the two trend lines approximating linear parts of the TGA curve before and during decomposition.
Density functional theory (DFT) calculations for the periodic systems were carried out using CASTEP v. 2017 (Biovia, San Diego, CA, USA) [25]. Several structural models of [(CH3)4P]BH4 were derived from the experimental data as described below. Generalized gradient approximation (GGA) was used with PBE functional and Tkatchenko–Scheffler dispersive correction [26]. The cut-off value of 600 eV was applied to achieve good energy convergence. The density of the k-point grid was set below 0.05 Å−1, and ultrasoft, generated on the fly pseudopotentials, were used to obtain more accurate lattice parameters. The unit cells were optimized under the given hydrostatic pressure using the BFGS algorithm. The Birch–Murnaghan equations of state (EoS) were fitted to the theoretical data using EoSFit software v. 7.60 [27,28].
3. Results and Discussion
3.1. Synthesis
Due to the low solubility of tetramethylphosphonium bromide in water and the decomposition of borohydrides occurring in the usual protic solvents, we attempted to carry out the mechanochemical synthesis of [(CH3)4P]BH4 according to Equation (1):
[(CH3)4P]Br + LiBH4 → [(CH3)4P]BH4 + LiBr
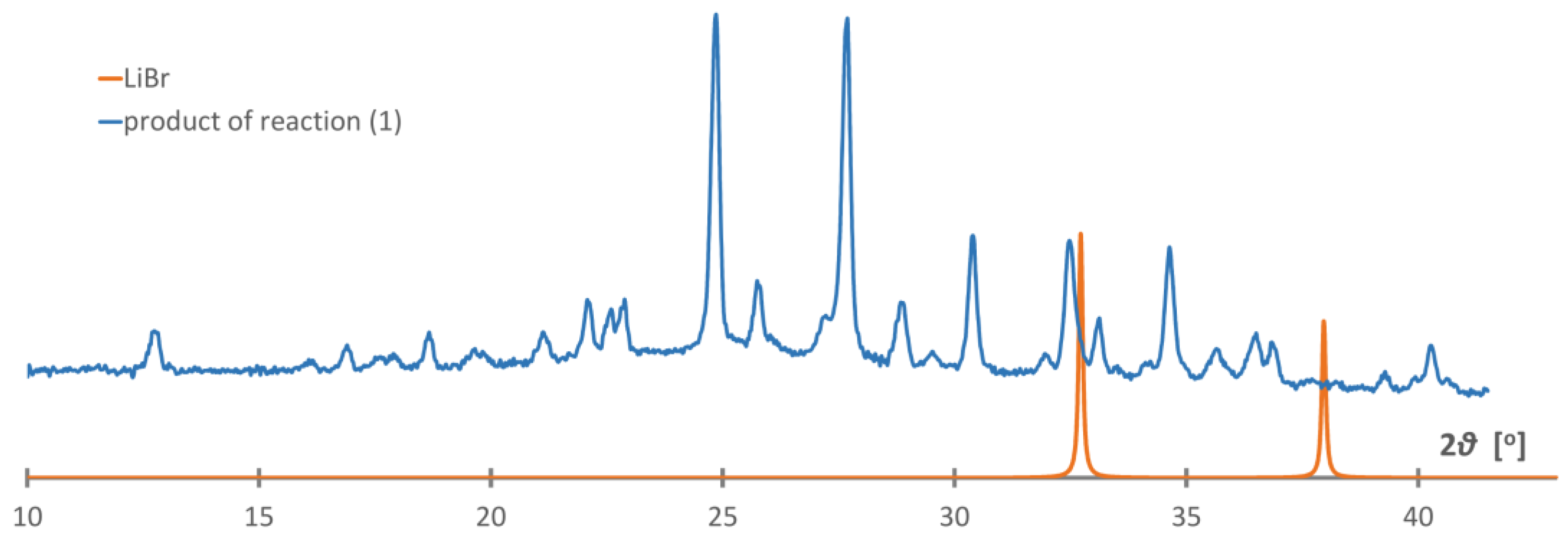
While the mechanochemical approach is very convenient for the study of a broad range of chemical compounds [29], it appeared that the reaction (1) did not occur according to our expectations, leading to unknown products and lacking the LiBr by-product in the post-reaction mixture, Figure 1. Apparently, the difference in lattice energies between the reagents and products may not be sufficient to perform this salt metathesis reaction without a solvent. As the diffraction pattern does not contain any signals of the reagents, an addition reaction may have occurred with the formation of mixed-ion salt. Unfortunately, the indexation of the PXD pattern appeared unsuccessful; therefore, we skipped further identification of this product.
As the mechanochemical approach appeared not to be applicable in this case, we utilized a more universal path of synthesis, allowing for the preparation of a broad range of ionic compounds [5]. First, the [(CH3)4P][Al(OC(CF3)3)4] precursor was prepared via the modified procedure for the synthesis of its ammonium analogue:
[(CH3)4P]Br + Li[Al(OC(CF3)3)4] → [(CH3)4P][Al(OC(CF3)3)4]↓ + LiBr
Subsequently, the precursor formed in reaction (2) was used for the salt metathesis reaction, leading to the desired product:
[(CH3)4P][Al(OC(CF3)3)4] + [(n-C4H9)4N]BH4 → [(CH3)4P]BH4↓ + [(n-C4H9)4N][Al(OC(CF3)3)4]
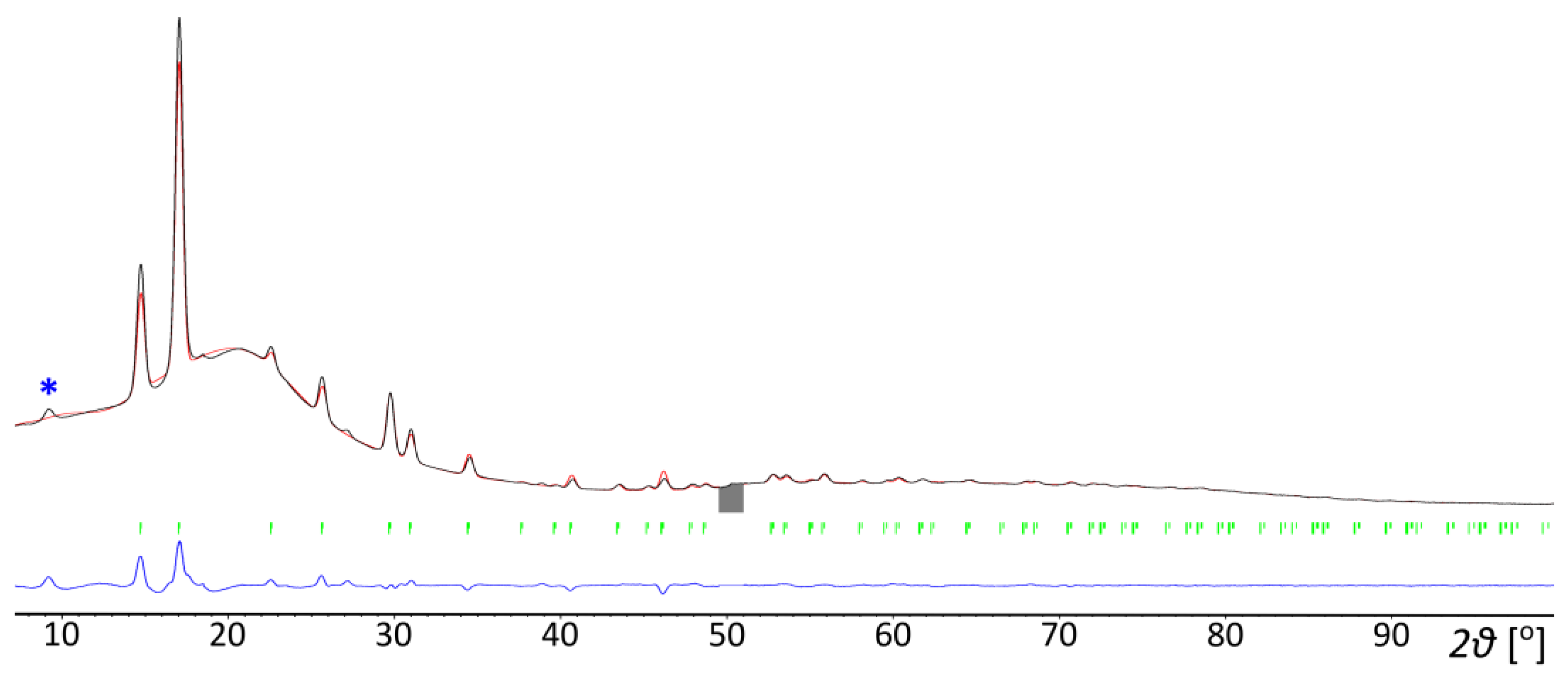
[(CH3)4P]BH4 was prepared in reaction (3) in a nearly pure form, with only barely detectable contamination with the [(n-C4H9)4N][Al(OC(CF3)3)4] by-product being visible on the PXD plot, Figure 2. Due to its very low solubility in dichloromethane, the product precipitated in microcrystalline form, and a few attempts of crystallization were necessary. The protic solvents or others, like liquid SO2 (in which a reduction to elemental sulfur occurred) appeared to be incompatible. Interestingly, the crystals grown from 1,2-dichloroethane appeared to be tetramethylphosphonium chloride, probably due to the contaminated solvent (we did not investigate this issue further). Finally, we were able to grow crystals from anhydrous acetonitrile, which appeared sufficiently stable in the reducing environment at room temperature.
3.2. Crystal Structures
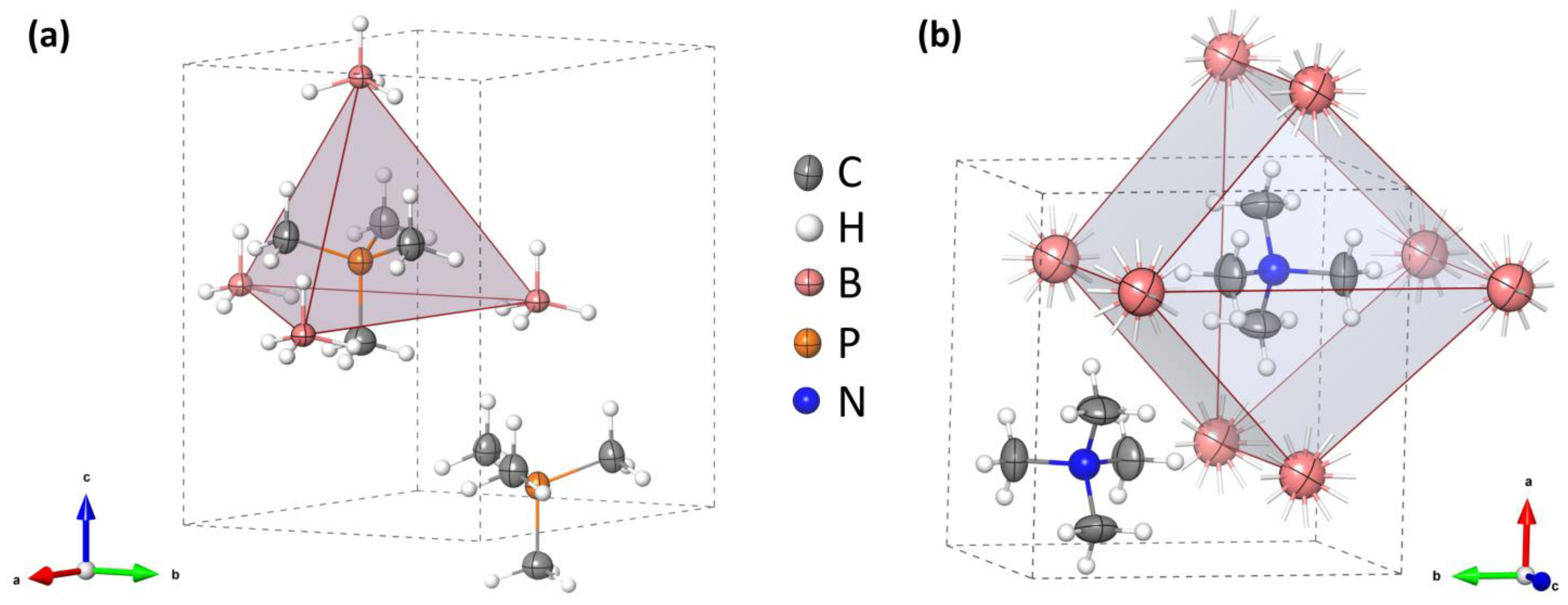
Tetramethylphosphonium borohydride crystallizes in a hexagonal unit cell with Z = 2, Table 1. The structure contains both cations and anions coordinated in a slightly distorted tetrahedral geometry, with P…B = 4.1240(11)–4.143(5) Å and B…P…B = 104.39(6)–114.04(5)°, and resembles the structure of wurtzite, Figure 3a. Such a crystal structure is adopted by several salts containing [(CH3)4P]+, [(CH3)4As]+, and [(CH3)4Sb]+ cations and monovalent anions, like O3−, Cl−–I−, BF4−, and PF6− [30,31,32]. On the other hand, the tetramethylammonium salts, [(CH3)4N]X, X = Cl–I, ClO4, BF4, and BH4, tend to crystallize in a tetragonal unit cell, in a distorted CsCl-type lattice [33,34,35], Figure 3b.
The structure of [(CH3)4P]BH4, despite the lack of disorder, is significantly more loosely packed than the disordered structure of [(CH3)4N]BH4. The molecular volume of the former is >25% larger (223.9 vs. 177.12 Å3 for [(CH3)4N]BH4 at room temperature), and its density remains ca. 6% lower despite its ca. 19% larger molecular mass. The low density of [(CH3)4P]BH4 is related to the voids of ca. 24 Å3 present in the structure (around 0 0 0.35 fractional coordinates, as listed by Olex2 [23]). Such voids might be sufficient to accommodate atoms like Ne [36]; however, no significant electron density peaks have been detected inside them.
Figure 3.
The crystal unit cell with ion coordination marked as a polyhedron for: (a) [(CH3)4P]BH4, P63mc (this work) and (b) [(CH3)4N]BH4, P4/nmm (the disordered hydrogen atoms were omitted for clarity) [35,37].

The nearest B–H…H–C contacts of ca. 2.2 Å are shorter than the sum of the van der Waals radii of hydrogen (2.4 Å). Due to the weak polarization of the C–Hδ+ bonds and the still substantial separation of the hydrogen atoms, these dihydrogen interactions must be of rather moderate strength. However, they are still able to fix the positions of the three hydrogen atoms of the borohydride group to remain closest to the respective hydrogen atoms of the methyl groups [38]. This is a different situation than in the case of [(CH3)4N]BH4, where the dihydrogen interactions with the disordered hydrogen atoms of BH4− anions are clearly much weaker (H…H = 2.47(3) Å; a slightly shorter H…H distance of 2.40(2) Å involves the ordered H atom of the BH4− moiety). Although the H…H contacts are shorter for [(CH3)4P]BH4, the distance between the adjacent heavy atoms, B…Cmin, is smaller in its ammonium analogue: 3.752(7) Å at room temperature [35], as compared to 3.833(4) Å at 100 K and 3.91(3) Å at 300 K. Obviously, the tetragonal structure of [(CH3)4N]BH4 facilitates better separation of B–H…H–C.
The crystal structure of [(CH3)4P]Cl shares its most important features with that of analogous borohydride, Table 1, and will not be discussed in detail. It may be noticed that the chloride expands slightly less than the borohydride when the temperature rises from 100 K to 300 K, 3.4% vs. 4.5%, respectively (a = 6.8779(2), c = 9.5941(4), V/Z = 196.53 Å3 for [(CH3)4P]Cl at 300 K).
3.3. Thermal Decomposition
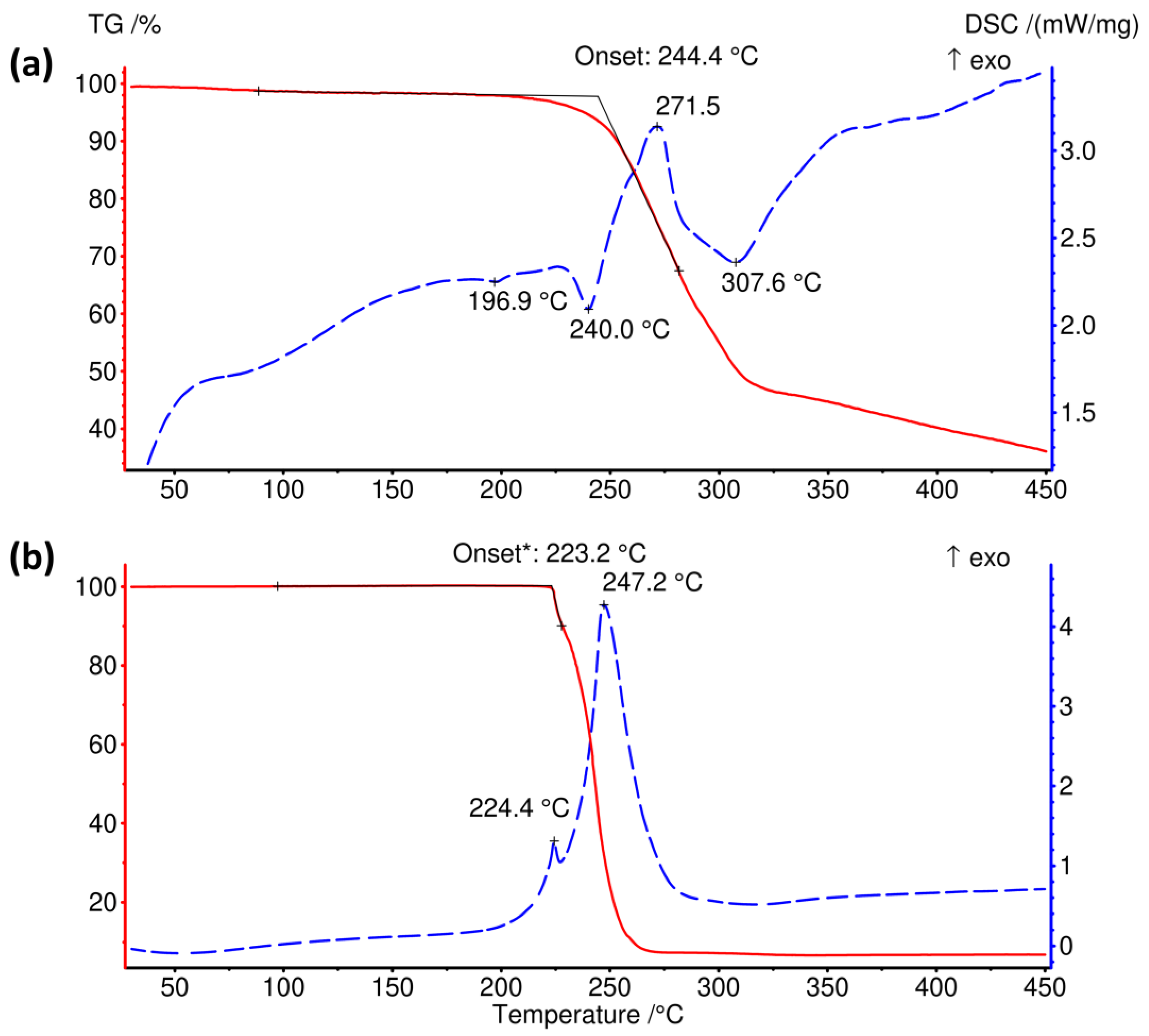
The thermal decomposition of [(CH3)4P]BH4 is preceded by an endothermic event on the DSC curve around 197 °C corresponding to 0.14 kJ mol−1, Figure 4. Such low heat indicates that this could be the result of a polymorphic transition or an artefact introduced by an unidentified minor contaminant.
The onset of mass loss occurs simultaneously with an endothermic DSC peak at 240 °C (1.43 kJ mol−1). The thermal decomposition process involves a few endo- and exothermic steps; some of them partially overlap, which is reflected by the complicated shape of the DSC curve, Figure 4a. The thermal effect of the exothermic peak at ca. 272 °C may be estimated as −8.05 kJ mol−1, while that of the endothermic peak at 308 °C—is 9.57 kJ mol−1. Although the mass loss rate significantly decreases above 320 °C, decomposition is clearly not terminated till the highest temperature of 450 °C, with the residual mass of 37.3 wt%.
The ammonium analogue of [(CH3)4P]BH4 decomposes in a different fashion, Figure 4b. In the case of [(CH3)4N]BH4, thermal decomposition occurs above ca. 223 °C in the two overlapping exclusively exothermic steps (ca. 0.7 and 53.1 kJ mol−1). The TGA curve flattens rather sharply above 270 °C, leaving the residual mass of only ca. 7 wt%.
3.4. High-Pressure Evolution of [(CH3)4P]BH4 as Probed by Raman Spectroscopy and DFT
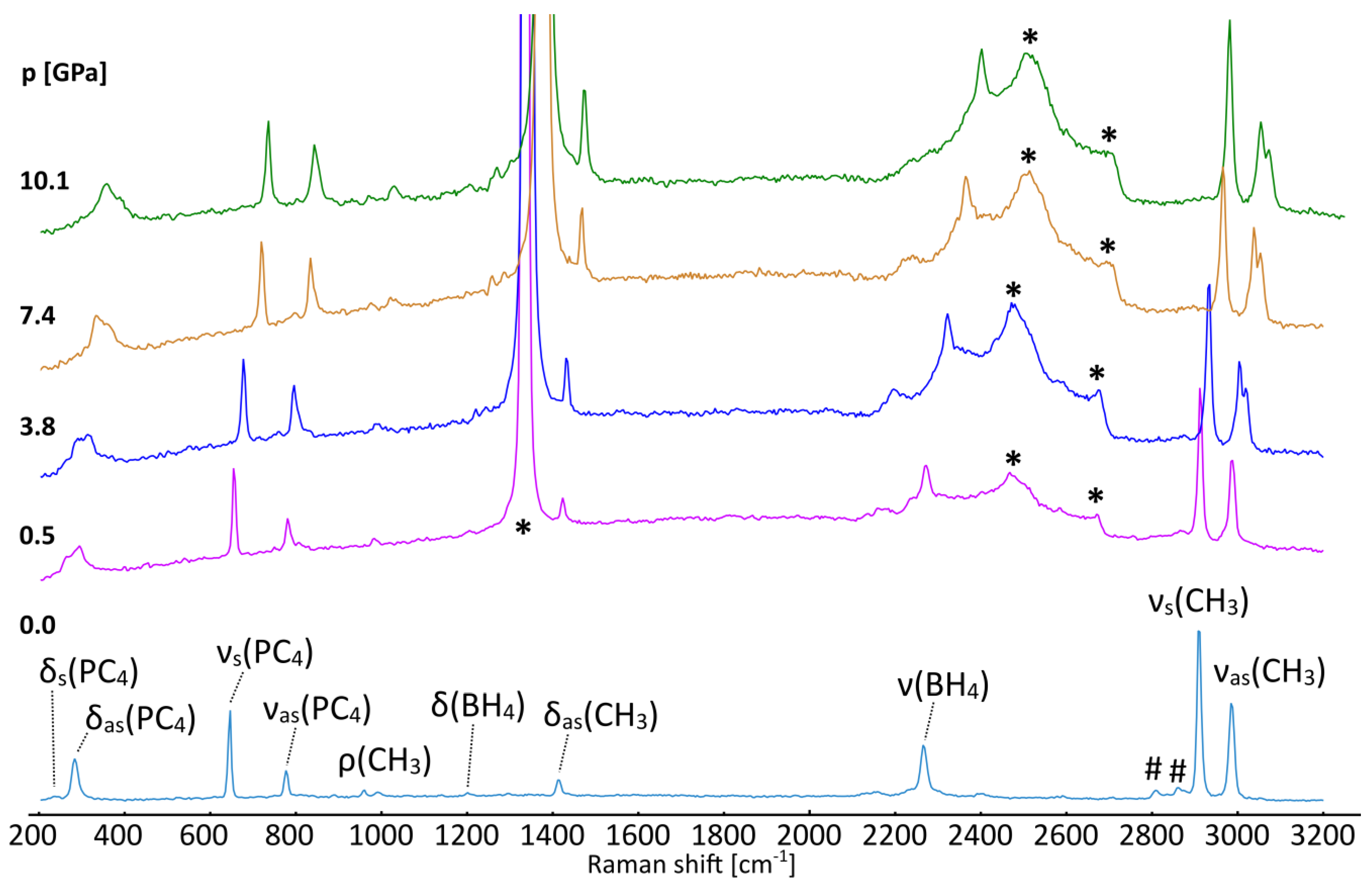
The Raman spectrum recorded for the recrystallized sample of [(CH3)4P]BH4 placed in a capillary confirms its purity, Figure 5 (bottom). No significant signals from the [(n-C4H9)4][Al(OC(CF3)3)4] by-product or related alkoxyaluminates were detected [18,19]. The spectrum looks as expected for the investigated compound, with it sharing the features of borohydride and tetramethylphosphonium moieties [23,37,39,40].
Under compression in a diamond anvil cell, the spectra reveal a distinct change between 0.53 and 1.86 GPa. The high-frequency νas(CH3) mode splits, resulting in the two signals above 2990 cm−1, Figure 5. This change is not accompanied by any noticeable split of the other Raman peaks, which might also be biased by the limited quality of the spectra measured for the sample loaded into the DAC. A similar divergence of this mode has been reported for [(CH3)4N]BH4 around 5 GPa, which takes place simultaneously with a more subtle split of the δ(CH3) mode [37]. These changes are related to the ordering of BH4− anions in the P4/nmm structure and the formation of the P21212 phase, resembling the low-temperature behavior of [(CH3)4N]ClO4 [41].
The lack of significant alterations in the PC4 and BH4 bands indicates that the pressure-induced recombination of the hydridic and protic hydrogen atoms does not occur up to ca. 10 GPa. Such a process would probably require much larger pressure to achieve more pronounced compression of the B–H…H–C contacts.
To gain a better insight into the possible nature of the observed phase transition, we conducted DFT calculations for several models of [(CH3)4P]BH4. Besides the P63mc unit cell, the structures proposed for [(CH3)4N]BH4 were considered. As has been mentioned above, the ambient pressure crystal structure of the latter compound (of P4/nmm symmetry) reveals disorder of the BH4− groups, cf. Figure 3, and cannot be directly optimized [35]. Therefore the two structures reported for [(CH3)4N]BH4 above 5 GPa (of P21212 symmetry) and above 20 GPa (of P-421m symmetry) were used for the calculations [37]. The selected parameters computed for the models considered here are presented in Figure 6, while the numerical data is contained in the Supplementary Materials (Table S1 and CIF files).
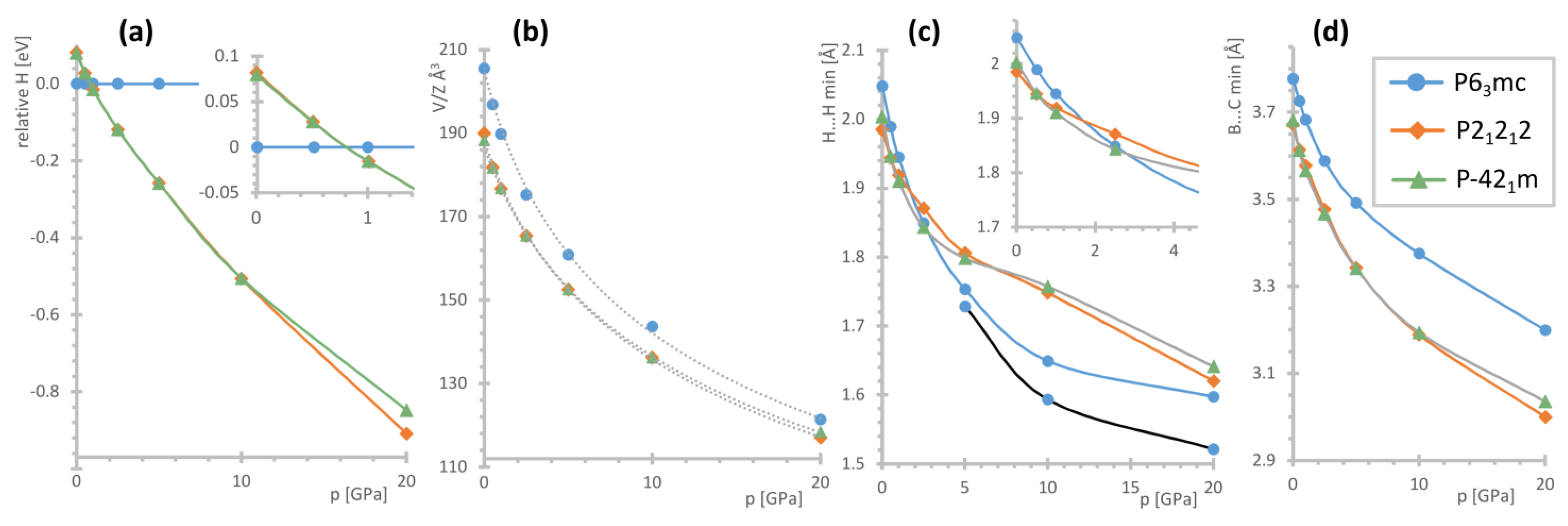
According to our DFT results, the P63mc structure is the most stable at ambient pressure by ca. 0.08 eV (i.e., 7.7 kJ mol−1) as compared to the other models considered. The unit cell optimized with the use of dispersive corrections reveals ca. −4.1% discrepancy from the experimentally determined volume at 100 K (and ca. −8.2% from the V/Z at 300 K), which falls rather within the values expected for the accuracy of such computational methods. The optimized nearest H…H contact of 2.048 Å is 7.7% shorter than those refined to the experimental data; at the same time the computed B…C minimal distance of 3.777 Å is only ca. 1.5% shorter than the experimental one (3.833(4) Å at 100 K).
The P21212 and P-421m structures of considerably better packing (ca. 8% smaller molecular volume) already become energetically favored above 0.8 GPa, Figure 6a. Both these structures converge to similar geometries and are energetically indistinguishable (energy difference of around 0–2 meV) besides the highest computed pressure point, where the lower symmetry constraints of the P21212 allow for more significant lowering of its energy. The range of pressure required for a polymorphic transition, as indicated by theoretical evidence, remains consistent with the high-pressure Raman investigation.
The theoretical high-pressure data were fitted to the third-order Birch–Murnaghan equation of state (EoS), Table 2. As could be expected, the P63mc phase is the most compressible, although all the obtained bulk moduli remain within the typical values reported for borohydrides [36,42,43]. While the reported bulk modulus of [(CH3)4N]BH4 is significantly lower (5.9(6) GPa), it must be mentioned that this number was calculated for the equation optimized with a relatively high value of B0′ = 9.6(4). At the same time, it can be noticed that this material still shows rather similar compressibility (ca. 64% vs. 59–63% predicted for various phases of [(CH3)4P]BH4 within the 0–20 GPa range) [37].
Interestingly, the H…H shortest contacts and the distances between the adjacent heavy atoms, mainly B…C, evolve in a different way. The latter shows similar behavior for all the phases, with it remaining the largest for the P63mc structure within the whole pressure range studied, Figure 6d. However, while the dihydrogen contacts for the P63mc phase are the longest at the lowest pressure range, they shorten the most among all the models considered during compression to 5 GPa. Moreover, in this case, the C–H…H–C contacts become even shorter, Figure 6c. Consequently—it could be expected that such geometry would not be favored energetically under high pressure.
4. Conclusions
Tetramethylphosphonium borohydride may be successfully prepared via general ion metathesis according to Equations (2) and (3). Although the convenience of this method lies in a lack of necessity for its optimization, it requires the performance of a two-step process and the utilization of not broadly available chemicals. It appears that due to the hampered reactivity towards acetonitrile observed during the crystallization attempts, a simple, one-step reaction similar to Equation (1) might be proposed for the preparation of this compound. This remains beyond the scope of the current report, however.
[(CH3)4P]BH4 decomposes thermally in several endo- and exothermic steps, in strong contrast with [(CH3)4N]BH4, for which solely exothermic processes are observed. The onset of the mass loss of ca. 20 °C higher also indicates the slightly higher kinetic stability of the former compound.
Although [(CH3)4P]BH4 crystallizes in a different structure than its ammonium analogue, our experimental and theoretical results indicate that they may share the same polymorph (either the P21212 or P-421m structure of the related salts). One of the factors rendering the P63mc structure relatively unstable is the proximity of the C–H…H–C contacts, which becomes unavoidable under larger pressure due to the crystallographic symmetry.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/ma16155334/s1. Table S1: The results of the DFT calculations for [(CH3)4P]BH4; the optimized structures of [(CH3)4P]BH4 at several pressure points.
Funding
The support from the National Science Centre (Poland), grant Sonata: UMO-2016/21/D/ST5/01966, is greatly appreciated. The authors thank the Biopolymers Laboratory, Faculty of Physics, University of Warsaw, for the access to the Agilent Supernova X-ray single-crystal diffractometer, co-financed by the European Union within the ERDF Project POIG.02.01.00-14-122/09.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The supplementary crystallographic data for this paper can be obtained free of charge from the Cambridge Crystallographic Data Centre: CCDC entries 2271774–2271777. The raw data from DFT calculations are contained in the Supplementary Materials. Other data may be provided on request by the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of the data; in the writing of the manuscript; or in the decision to publish the results.
References
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Černý, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacher, F.; Jensen, T.R. Metal Borohydrides and Derivatives—Synthesis, Structure and Properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef]
- Suárez-Alcántara, K.; García, J.R.T. Metal Borohydrides beyond Groups i and Ii: A Review. Materials 2021, 14, 2561. [Google Scholar] [CrossRef]
- DOE US. Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles. US Drive 2017, 1, 1–29. [Google Scholar]
- Hagemann, H.; Černý, R. Synthetic Approaches to Inorganic Borohydrides. Dalt. Trans. 2010, 39, 6006. [Google Scholar] [CrossRef]
- Jaroń, T.; Orłowski, P.A.; Wegner, W.; Fijałkowski, K.J.; Leszczyński, P.J.; Grochala, W. Hydrogen Storage Materials: Room-Temperature Wet-Chemistry Approach toward Mixed-Metal Borohydrides. Angew. Chem. Int. Ed. 2015, 54, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Starobrat, A.; Tyszkiewicz, M.J.; Wegner, W.; Pancerz, D.; Orłowski, P.A.; Leszczyński, P.J.; Fijalkowski, K.J.; Jaroń, T.; Grochala, W. Salts of Highly Fluorinated Weakly Coordinating Anions as Versatile Precursors towards Hydrogen Storage Materials. Dalt. Trans. 2015, 44, 19469–19477. [Google Scholar] [CrossRef]
- Wegner, W.; Jaroń, T. Synthesis, Polymorphism and Thermal Decomposition Process of (N-C4H9)4NRE(BH4)4 for RE = Ho, Tm and Yb. Materials 2021, 14, 1329. [Google Scholar] [CrossRef]
- Wegner, W.; Fijalkowski, K.J. Synthesis Method of Unsolvated Organic Derivatives of Metal Borohydrides. Materials 2022, 15, 8653. [Google Scholar] [CrossRef] [PubMed]
- Raber, D.J.; Guida, W.C. Tetrabutylammonium Borohydride. Borohydride Reductions in Dichloromethane. J. Org. Chem. 1976, 41, 690–696. [Google Scholar] [CrossRef]
- Fraser, K.J.; MacFarlane, D.R. Phosphonium-Based Ionic Liquids: An Overview. Aust. J. Chem. 2009, 62, 309–321. [Google Scholar] [CrossRef]
- Rios, A.; O’Donoghue, A.M.C.; Amyes, T.L.; Richard, J.P. Formation and Stability of Organic Zwitterions—The Carbon Acid PK as of the Trimethylsulfonium and Tetramethylphosphonium Cations in Water. Can. J. Chem. 2005, 83, 1536–1542. [Google Scholar] [CrossRef]
- Saidov, B.I.; Borisov, A.P.; Makhaev, V.D.; Bojko, G.N.; Antsyshkina, A.S.; Kedrova, N.S.; Mal’tseva, N.N. Complexes of Zinc Tetrahydroborate with Tetrahydroborates of Organic Cations. Russ. J. Inorg. Chem. 1990, 35, 353–357. [Google Scholar]
- Antsyshkina, A.S.; Sadikov, G.G.; Borisov, P.; Makhaev, V.D. Complexes of Yttrium, Thulium, and Lutetium Tetrahydridoborates with Tetraphenylphosphonium Tetrahydridoborate (Ph4P)[M(BH4)4] (M = Y, Tm, Lu): Crystal Structure of (Ph4P)[Tm(BH4)4]. Russ. J. Inorg. Chem. 2001, 46, 1141–1146. [Google Scholar]
- Heal, H.G. “Onium” Base Borohydrides-II. Some Phenyl Compounds. J. Inorg. Nucl. Chem. 1961, 16, 208–212. [Google Scholar] [CrossRef]
- Cao, W.; Zhou, J.; Kochovski, Z.; Miao, H.; Gao, Z.; Sun, J.K.; Yuan, J. Ionic Organic Cage-Encapsulated Metal Clusters for Switchable Catalysis. Cell Rep. Phys. Sci. 2021, 2, 100546. [Google Scholar] [CrossRef]
- Yang, X.; Ullah, Z.; Stoddart, J.F.; Yavuz, C.T. Porous Organic Cages. Chem. Rev. 2023, 123, 4602–4634. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Q. Encapsulating Metal Nanocatalysts within Porous Organic Hosts. Trends Chem. 2020, 2, 214–226. [Google Scholar] [CrossRef]
- Raabe, I.; Wagner, K.; Guttsche, K.; Wang, M.; Grätzel, M.; Santiso-Quiñones, G.; Krossing, I. Tetraalkylammonium Salts of Weakly Coordinating Aluminates: Ionic Liquids, Materials for Electrochemical Applications and Useful Compounds for Anion Investigation. Chem.—Eur. J. 2009, 15, 1966–1976. [Google Scholar] [CrossRef]
- Malinowski, P.J.; Jaroń, T.; Domańska, M.; Slattery, J.M.; Schmitt, M.; Krossing, I. Building Blocks for the Chemistry of Perfluorinated Alkoxyaluminates [Al{OC(CF3)3}4]−: Simplified Preparation and Characterization of Li+-Cs+, Ag+, NH4+, N2H5+ and N2H7+ salts. Dalt. Trans. 2020, 49, 7766–7773. [Google Scholar] [CrossRef]
- Krossing, I.; Reisinger, A. Chemistry with Weakly-Coordinating Fluorinated Alkoxyaluminate Anions: Gas Phase Cations in Condensed Phases? Coord. Chem. Rev. 2006, 250, 2721–2744. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Oxfordshire, UK, 2014. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Petricek, V.; Dusek, M.; Palatinus, L.; Petříček, V.; Dušek, M.; Palatinus, L.; Petrícek, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General Features. Z. Krist. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, R.J.; Gonzalez-Platas, J.; Alvaro, M. EosFit7c and a Fortran Module (Library) for Equation of State Calculations. Z. Krist. 2014, 229, 405–419. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R. EosFit7-GUI: A New Graphical User Interface for Equation of State Calculations, Analyses and Teaching. J. Appl. Crystallogr. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Ravnsbaek, D.B.; Sørensen, L.H.; Filinchuk, Y.; Besenbacher, F.; Jensen, T.R. Screening of Metal Borohydrides by Mechanochemistry and Diffraction. Angew. Chem. Int. Ed. 2012, 51, 3582–3586. [Google Scholar] [CrossRef]
- Collins, E.; June Sutor, D.; Mann, F.G. The Crystal Structure of Tetramethylarsonium Bromide. J. Chem. Soc. 1963, 4130–4145. [Google Scholar] [CrossRef]
- Burbach, G.; Dou, S.-Q.; Weiss, A. Molecular Motion in Solid [Sb(CH3)4]X, X = Cl, Br, I, PF6, and BF4. 1H, 19F NMR Second Moment and Crystal Structure. Berichte Bunsenges. Phys. Chem. 1989, 93, 1302–1309. [Google Scholar] [CrossRef]
- Assenmacher, W.; Jansen, M. Zur Kenntnis Der Ionischen Ozonide P(CH3)4O3 Und As(CH3)4O3. ZAAC—J. Inorg. Gen. Chem. 1995, 621, 431–434. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G., IV. The Crystal Structure of the Tetramethyl Ammonium Halides. Z. Krist.—Cryst. Mater. 1928, 67, 91–105. [Google Scholar] [CrossRef]
- Evans, D.J.; Hughes, D.L. Structure of Tetramethylammonium Bromide: A Redetermination. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1990, 46, 1452–1454. [Google Scholar] [CrossRef]
- Jaroń, T.; Grochala, W. Tetra-Methyl-Ammonium Borohydride from Powder Data. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o2171. [Google Scholar] [CrossRef]
- Jaroń, T.; Starobrat, A.; Struzhkin, V.V.; Grochala, W. Inclusion of Neon into Yttrium Borohydride Structure at Elevated Pressure—An Experimental and Theoretical Study. Eur. J. Inorg. Chem. 2020, 2020, 3846–3851. [Google Scholar] [CrossRef]
- Dalton, D.A.; Somayazulu, M.; Goncharov, A.F.; Hemley, R.J. Static Compression of Tetramethylammonium Borohydride. J. Phys. Chem. A 2011, 115, 11033–11038. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Pérez, E.J.; Núñez, R.; Viñas, C.; Sillanpää, R.; Teixidor, F. The Role of C-H⋯H-B Interactions in Establishing Rotamer Configurations in Metallabis(Dicarbollide) Systems. Eur. J. Inorg. Chem. 2010, 2010, 2385–2392. [Google Scholar] [CrossRef]
- Kornath, A.; Blecher, O.; Neumann, F.; Ludwig, R. Vibrational Spectra of the Tetramethylpnikogenonium Ions. J. Mol. Spectrosc. 2003, 219, 170–174. [Google Scholar] [CrossRef]
- Dietzel, P.D.C.; Kremer, R.K.; Jansen, M. Superoxide Compounds of the Large Pseudo-Alkali-Metal Ions Tetramethylammonium, -Phosphonium, and -Arsonium. Chem.—Asian J. 2007, 2, 66–75. [Google Scholar] [CrossRef]
- Palacios, E.; Burriel, R.; Ferloni, P. The Phases of [(CH3)4N](ClO4) at Low Temperature. Acta Crystallogr. Sect. B Struct. Sci. 2003, 59, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Filinchuk, Y.; Richter, B.; Jensen, T.R.; Dmitriev, V.; Chernyshov, D.; Hagemann, H. Porous and Dense Magnesium Borohydride Frameworks: Synthesis, Stability, and Reversible Absorption of Guest Species. Angew. Chem. Int. Ed. 2011, 50, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, N.A.; Roedern, E.; Łodziana, Z.; Nielsen, D.B.; Jensen, T.R.; Talyzin, A.V.; Černý, R.; Chernyshov, D.; Dmitriev, V.; Palasyuk, T.; et al. High-Pressure Study of Mn(BH4)2 Reveals a Stable Polymorph with High Hydrogen Density. Chem. Mater. 2016, 28, 274–283. [Google Scholar] [CrossRef]
Figure 1.
PXD pattern of the products of the reaction attempted according to Equation (1), blue curve, compared to the pattern generated for LiBr, orange curve (Co Kα).
Figure 1.
PXD pattern of the products of the reaction attempted according to Equation (1), blue curve, compared to the pattern generated for LiBr, orange curve (Co Kα).

Figure 2.
Rietveld refinement profile of [(CH3)4P]BH4 prepared in reaction (3). The strongest signal of the [(n-C4H9)4N][Al(OC(CF3)3)4] by-product has been marked with an asterisk. Cu Kα radiation, 150 K. a = 6.9518(4) Å, c = 10.4183(8) Å, V = 436.036 Å3, wRp = 10.86%. Black line—experimentally observed data, red line—calculated data, blue line—difference between the observed and calculated data.
Figure 2.
Rietveld refinement profile of [(CH3)4P]BH4 prepared in reaction (3). The strongest signal of the [(n-C4H9)4N][Al(OC(CF3)3)4] by-product has been marked with an asterisk. Cu Kα radiation, 150 K. a = 6.9518(4) Å, c = 10.4183(8) Å, V = 436.036 Å3, wRp = 10.86%. Black line—experimentally observed data, red line—calculated data, blue line—difference between the observed and calculated data.

Figure 4.
Comparison of the TGA and DSC curves of (a) [(CH3)4P]BH4 and (b) [(CH3)4N]BH4. *—onset of the first of two overlapping decomposition stages. The arrow points at the exothermic direction of DSC curves.
Figure 4.
Comparison of the TGA and DSC curves of (a) [(CH3)4P]BH4 and (b) [(CH3)4N]BH4. *—onset of the first of two overlapping decomposition stages. The arrow points at the exothermic direction of DSC curves.

Figure 5.
Raman spectra of [(CH3)4P]BH4 crystallized from acetonitrile at ambient (bottom) and elevated pressures (as listed in GPa). Identification of the most prominent vibrational modes has been shown for the ambient-pressure spectra on the basis of refs. [37,39,40]. *—the most intense signals from the diamond; #—overtones and combinatorial modes.
Figure 5.
Raman spectra of [(CH3)4P]BH4 crystallized from acetonitrile at ambient (bottom) and elevated pressures (as listed in GPa). Identification of the most prominent vibrational modes has been shown for the ambient-pressure spectra on the basis of refs. [37,39,40]. *—the most intense signals from the diamond; #—overtones and combinatorial modes.

Figure 6.
Summary of the DFT calculations for the specified structural models of [(CH3)4P]BH4: (a) relative enthalpies, (b) molecular volumes (points) and their fits to the Birch–Murnaghan equation (gray dotted lines), (c) the shortest B–H…H–C contacts; for the P63mc phase, the C–H…H–C contacts are also shown using a black line; and (d) the shortest B…C contacts. The low-pressure regions were expanded for (a,c) for better visibility.
Figure 6.
Summary of the DFT calculations for the specified structural models of [(CH3)4P]BH4: (a) relative enthalpies, (b) molecular volumes (points) and their fits to the Birch–Murnaghan equation (gray dotted lines), (c) the shortest B–H…H–C contacts; for the P63mc phase, the C–H…H–C contacts are also shown using a black line; and (d) the shortest B…C contacts. The low-pressure regions were expanded for (a,c) for better visibility.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic data for the structures reported in this work.
| Compound | Tetramethylphosphonium Borohydride | Tetramethylphosphonium Chloride | |
|---|---|---|---|
| Formula | [(CH3)4P]BH4 | [(CH3)4P]Cl | |
| T [K] | 100 | 300 | 100 |
| Space group | P63mc | P63mc | P63mc |
| Unit cell dimensions [Å3] | a = 6.9190(2) c = 10.3350(3) | a = 7.0231(17) c = 10.483(3) | a = 6.8427(2) c = 9.3740(3) |
| c/a | 1.494 | 1.493 | 1.370 |
| V [Å3] | 428.48(2) | 447.8(2) | 380.11(2) |
| Z | 2 | 2 | 2 |
| V/Z [Å3] | 214.24 | 223.9 | 190.06 |
| d [g cm−3] | 0.821 | 0.786 | 1.106 |
| (P–C)min [Å] | 1.781(3) | 1.73(3) | 1.777(2) |
| (P–C)max [Å] | 1.789(7) | 1.761(14) | 1.790(6) |
| (C–P–C)min [°] | 109.41(12) | 108.1(6) | 108.93(9) |
| (C–P–C)max [°] | 109.54(12) | 110.8(6) | 110.01(9) |
| (B–H)min [Å] | 1.186(10) | 1.190(10) | - |
| (B–H)max [Å] | 1.190(10) | 1.190(10) | - |
| (C–H)min [Å] | 1.079(10) | 1.082(10) | 1.061(7) |
| (C–H)max [Å] | 1.081(10) | 1.083(10) | 1.071(8) |
| (P…An)min [Å] * | 4.1240(11) | 4.18(2) | 3.8242(9) |
| (P…P)min [Å] | 6.53151(14) | 6.6268(14) | 6.12988(14) |
| (An…An)min [Å] * | 6.53151(14) | 6.6268(14) | 6.12988(14) |
| (H…H)min [Å] | 2.22(4) | 2.16(13) | 2.71(3) |
| Goodness of fit | 1.087 | 1.134 | 1.046 |
| R1 [%] | 3.77 | 9.20 | 2.81 |
| wR2 [%] | 10.13 | 30.33 | 7.85 |
| CSD No | 2271776 | 2271774 | 2271775 |
* Distance involving the center of the relevant anion [An].
Table 2.
Summary of the parameters of the Birch–Murnaghan EoS obtained for the theoretical data. The estimated standard deviations are given in parentheses. B0′ was refined.
Table 2.
Summary of the parameters of the Birch–Murnaghan EoS obtained for the theoretical data. The estimated standard deviations are given in parentheses. B0′ was refined.
| Phase of [(CH3)4P]BH4 | V0/Z [Å3] | B0 [GPa] | B0′ | Max Δp [GPa] |
|---|---|---|---|---|
| P63mc | 205.1(22) | 12.3(6) | 4.3 | 0.55 |
| P21212 | 188.4(7) | 15.0(3) | 4.3 | 0.18 |
| P-421m | 186.6(5) | 16.2(2) | 4.3 | 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jaroń, T. The Preparation, Characterization, and Pressure-Influenced Dihydrogen Interactions of Tetramethylphosphonium Borohydride. Materials 2023, 16, 5334. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16155334
AMA Style
Jaroń T. The Preparation, Characterization, and Pressure-Influenced Dihydrogen Interactions of Tetramethylphosphonium Borohydride. Materials. 2023; 16(15):5334. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16155334
Chicago/Turabian StyleJaroń, Tomasz. 2023. "The Preparation, Characterization, and Pressure-Influenced Dihydrogen Interactions of Tetramethylphosphonium Borohydride" Materials 16, no. 15: 5334. https://0-doi-org.brum.beds.ac.uk/10.3390/ma16155334
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.